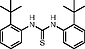Subscribe to RSS
DOI: 10.1055/s-0039-1690338
Scalable Synthesis of 1,3,4,5-Tetraaryl Imidazolium Salts as Precursors of Sterically Demanding N-Heterocyclic Carbenes
Publication History
Received: 05 November 2019
Accepted after revision: 15 November 2019
Publication Date:
23 January 2020 (online)

Abstract
A convenient, large-scale, and cost-efficient synthesis of 4,5-diarylsubstituted N,N-diarylimidazolium salts is described. A variety of 1,3,4,5-tetraaryl imidazolium salts with increasing electron donation and steric bulk of the N-aryl groups was synthesized in good yields. In the key step, readily available N,N′-diarylthioureas and benzoin/anisoin are coupled to give imidazole-2-thiones, followed by imidazolium salt formation by oxidative desulfurization. In this way, N,N-diarylimidazolium salts with 2-methoxy, 2-methyl, and 2-isopropyl substituents could be obtained; the synthesis of their 2-tert-butyl, 2,6-dimethyl, and 2,6-diisopropyl analogues failed.
#
N-Heterocyclic carbenes (NHCs) have attracted considerable attention as alternatives for widely used phosphine complexes in homogeneous catalysis.[3] Among them, sterically demanding and electron-rich carbenes have been successfully utilized in most catalytic transformations. Particularly, QSAR analysis shows that imidazolylidene ligands with the backbone carbon atoms substituted by phenyl groups most efficiently promote catalytic activity of second-generation Ru catalysts in olefin metathesis.[4] As a part of our efforts to provide access to effective metathesis catalysts, we were interested in a cost-effective and easily scalable synthesis of 1,3,4,5-tetraaryl imidazolium salts.[5] Especially, we are seeking methods and procedures that are applicable industrially, avoiding chromatographic separation.
Over the past decades, many synthetic methods for the preparation of imidazolium salts have been reported.[6] [7] Although, the first synthesis of a tetraarylimidazolium salt was established almost 50 years ago, examples of 4,5-diaryl-substituted imidazolium salts are rare.[8] Recently, one highly sterically demanding example (Ar = Mes) was reported (Scheme [1], A).[8d] Most recently, an aldimine coupling followed by cyclization with formaldehyde was used for the preparation of a wide variety of 4,5-diaryl-substituted NHCs with para-substituted N-phenyl groups (Scheme [1], B).[8e] [f] An interesting method for formation of 1,3,4,5-tetraarylimidazoles is reaction of benzyl dianil with sodium in diethyl ether followed by addition of carbon disulfide (Scheme [1], C).[8g]

Unfortunately, protocols A and C are not really scalable and protocol B is restricted to the preparation of imidazolium salts with para-substituents on the N-aryl groups. The strategy outlined in Scheme [2] appears to be the most convenient synthetic route for the large-scale and cost-efficient preparation of 4,5-diaryl-substituted imidazolium salts with sterically demanding N-aryl groups.

A number of synthetic protocols to access 1,3-disubstituted thioureas 1 have been reported.[9] Past approaches include reaction of thiophosgene,[10] isothiocyanates,[11] and 1,1′-thiocarbonyldiimidazole[12] with amines. Recently, the direct conversion of aniline and CS2 into thiourea 1 has been developed.[9b] [13] After initial experiments, applying different published approaches, we found that N,N-diarylthioureas 1c–i and 1m could be generated in good yields by the reaction of the amine with the CS2 in water; whereas heating under reflux for 6 h in diglyme was required for the formation of thioureas 1j and 1k bearing isopropyl- and tert-butyl groups at the ortho-positions, respectively. Furthermore, thioureas 1a, 1b, and 1n were obtained in better yields by using the latter method. The substitution patterns of the N-aryl groups were selected on the basis of availability of the corresponding anilines on large scale, including electron-rich (c, d, e) and with increased steric demand (g–k, m, n) substrates.
It should be noted at this point that only benzoin and anisoin are commercially available; therefore, no more benzoins were used in this survey. To introduce other substitution patterns into the aryl-groups at positions 4 and 5 requires significant synthetic effort.[14]
Imidazolin-2-thiones were obtained in good to excellent yields by condensation of the N,N-diarylureas with benzoin or anisoin in acetic acid,[15] although the imidazolin-2-thione 2j, bearing an isopropyl-group at the ortho-position, was isolated in reduced yield. Imidazolin-2-thiones with ortho-substituents were obtained as a pair of diastereoisomers but because the products of the subsequent oxidation, imidazolium salts 3g–4i, do not exhibit atropoisomerism we did not separate the diastereoisomers. When the condensation was attempted with thioureas 1k, 1m, and 1n, no imidazolin-2-thiones were produced. Instead 2-[(2-tert-butylphenyl)amino]-1,2-diphenylethanone (5) was isolated (see Table [1] below). Attempts to achieve the cyclization in boiling hexanol in the presence of catalytic amounts of hydrochloric acid[16] were unsuccessful. It should be noted that attempts to form N,N-bis(2-tert-butylphenyl)-4,5-dimethylimidazolin-2-thione failed, as reported by Bach et al.[17] Interestingly, reaction of N,N′-bis(3-pyridinyl)thiourea 1l with benzoin under similar conditions in acetic acid gave 2-[(3-pyridyl)amino]-1,2-diphenylethanone (6) in excellent yield.
Oxidative desulfurization was performed using H2O2 in acetic acid. The initial products of oxidative desulfurization – the hydrogen sulfates[8b] [c] were obtained as viscous syrups after careful removal of acetic acid. Ion exchange with NaClO4 gave perchlorates 3a–j effectively as analytically pure compounds. A mixture of two diastereoisomers was formed in the case of ortho-isopropyl-substituted imidazolium salt 3j, as in the case of the 4,5-dimethyl analogue.[17] We expect that the carbene generated from 3j will not exhibit atropoisomerism because rotation around the N-Ar bond in the carbenes is not restricted.[17] Therefore, no further attempts to separate the diastereoisomers were undertaken. Ion exchange with BaCl2 furnished imidazolium chlorides 4a–i. Unfortunately, desulfurization of 2j and ion exchange with BaCl2 did not give a crystalline product; therefore, no further attempts to purify this chloride salt were undertaken.
In conclusion, we have reported a convenient, large-scale and cost-efficient synthesis of 4,5-diarylsubstituted imidazolium salts with electron-rich, and sterically demanding N-aryl groups (Table [1]). In this way, N,N-diarylimidazolium salts with ortho-methoxy, ortho-methyl, and ortho-isopropyl substituents can be obtained; whereas the synthesis of their 2-tert-butyl, 2,6-dimethyl, and 2,6-di-isopropyl analogues failed.
All starting materials and solvents were obtained from commercial suppliers and were used without further purification. 2-Methyl-5-tert-butylaniline was prepared according to the reported procedure.[18] Melting points of all synthesized compounds were determined in open capillaries and are uncorrected. NMR spectra were recorded with a Bruker Avance 400 (1H: 400 MHz, 13C: 101 MHz), Bruker Avance 600 (1H: 601 MHz, 13C: 151 MHz), or Jeol ECZ-600 R (1H: 600 MHz, 13C: 151 MHz) spectrometer; chemical shifts are given in δ ppm. IR spectra were recorded with a Bruker Tensor 27 spectrophotometer equipped with a ‘GoldenGate’ diamond ATR unit.
#
General Procedures for the Synthesis of Thioureas 1
General Procedure A: A stirred mixture of aniline (1 mol), sulfur (2 g), and potassium carbonate (2 g) in water (700 mL) was heated to 80 °C, carbon disulfide (0.6–1.0 mol) was added dropwise over 60 min and the mixture was then heated to reflux for 6 h. After cooling to ambient temperature, the precipitated product was filtered off, washed with 1 M hydrochloric acid, then with water, sucked as dry as possible and the crude product was recrystallized from boiling ethanol.
General Procedure B: A mixture of 2-isopropylaniline (135.0 g, 1 mol), sulfur (1 g), and potassium carbonate (2 g) in diglyme (1 L) was heated to 80 °C, then carbon disulfide (38 g, 0.5 mol) was added over 1 h. The mixture was then slowly heated to 180 °C (bath temperature) and stirred at this temperature for 6 h. The reaction mixture was then allowed to cool overnight to ambient temperature. The precipitate was filtered off with suction, washed with 1 M hydrochloric acid, then with water, sucked as dry as possible, washed with hexane and dried in vacuum.
#
General Procedure for the Synthesis of Imidazolin-2-thiones 2
Benzoin/anisoin (0.1 mol) and N,N′-diarylthiourea (0.1 mol) were heated to reflux in glacial acetic acid (150 mL) for 6–16 h and the mixture was then allowed to cool to ambient temperature. EtOH (50 mL) was added and the suspension was stirred in an ice bath to complete the precipitation. The precipitated product was filtered off with suction, washed with ethanol, then diethyl ether, and dried in vacuum.
#
General Procedure for the Synthesis of Perchlorates 3
Caution: Perchlorates are potentially explosive and should be treated as potentially hazardous compounds.
To a solution of imdiazolin-2-thione 2 (0.1 mol) in glacial acetic acid (200 mL), 35% aqueous H2O2 (34 mL, 0.4 mol) was added dropwise, allowing the temperature to rise to 50 °C, leading to a slightly turbid mixture that was subsequently stirred at ambient temperature for 4 h. All volatiles were removed by rotary evaporation ( Caution: Do not evaporate to dryness. When preparing larger volumes, it is critical to ensure that any excess of hydrogen peroxide is destroyed before workup; otherwise, explosive decomposition may occur). The residue was dissolved in MeOH (200 mL) and treated with a solution of NaClO4 (28.1 g, 0.2 mol) in a 2:1 (v/v) mixture of methanol/water (200 mL). A white solid precipitated upon additional stirring in an ice bath. The precipitate was filtered off and washed with water, diethyl ether and dried at r.t. in vacuum.
#
General Procedure for the Synthesis of Chlorides 4
Hydrogen peroxide (35%; 53 mL, 0.65 mol) was added dropwise to a stirred suspension of imidazolin-2-thione 2 (0.2 mol) in glacial acetic acid (200 mL). An exothermic reaction occurred with the reaction mixture reaching 60 °C at the end of the addition and the solution was stirred for a further 4 h. All volatiles were removed by rotary evaporation ( Caution: Do not evaporate to dryness. When preparing larger volumes, it is critical to ensure that any excess of hydrogen peroxide is destroyed before workup; otherwise, explosive decomposition may occur) and the residue was dissolved in MeOH (500 mL). A solution of BaCl2 (97.7 g, 0.4 mol) in water (200 mL) was added, and the suspension was filtered with suction through a G4 glass sinter covered with Celite®. The filtrate was evaporated and the residue was extracted with dichloromethane (3 × 100 mL). The combined organic layers were dried over MgSO4, filtered, and the filtrate evaporated. The residue was triturated with diethyl ether, the solid product was filtered with suction, washed with diethyl ether, and dried in vacuum.
#
N,N′-Bis(4-ethoxycarbonylphenyl)thiourea (1a)[19]
According to GP B, the reaction of ethyl 4-aminobenzoate (82.6 g, 0.5 mol) and carbon disulfide (45 mL, 0.75 mol) and recrystallization from boiling water afforded 1a. An analytically pure sample was obtained by recrystallization from ethanol.
Yield: 47% (44.1 g); mp 164–165 °C (Lit[19] 165 °C).
IR (ATR): 3284br, 1681s, 1591s, 1524s, 1507s, 1408s, 1328s, 1286s, 1229s, 1174s, 849s, 765s, 734s, 650s, 614s, 572s cm–1.
1H NMR (400 MHz, DMSO-d 6): δ = 10.33 (s, 1 H), 7.93 (d, J = 8.7 Hz, 2 H), 7.69 (d, J = 8.7 Hz, 2 H), 4.30 (q, J = 7.1 Hz, 2 H), 1.32 (t, J = 7.1 Hz, 3 H).
13C NMR (101 MHz, DMSO-d 6): δ = 179.09, 165.23, 143.71, 129.66, 125.04, 122.01, 60.44, 14.10.
Anal. Calcd for C19H20N2O4S: C, 61.27; H, 5.41; N, 7.52; S, 8.61. Found: C, 60.93; H, 5.38; N, 7.41; S, 8.72.
#
1,3-Bis(4-ethoxycarbonylphenyl)-4,5-diphenylimidazolin-2-thione (2a)
Reaction of thiourea 1a (44.0 g, 0.12 mol) and benzoin (25.1 g, 0.12 mol) in acetic acid (170 mL) for 6 h afforded 2a. An analytically pure sample was obtained by recrystallization from CHCl3/EtOH.
Yield: 76% (49.1 g); mp 305–307 °C (dec.).
IR (ATR): 1715s, 1606m, 1512m, 1336s, 1269s, 1172m, 1099s, 741s, 693s, 597s cm–1.
1H NMR (601 MHz, CDCl3): δ = 8.02–7.96 (m, 4 H), 7.42–7.35 (m, 4 H), 7.11 (t, J = 7.4 Hz, 2 H), 7.05 (t, J = 7.6 Hz, 4 H), 6.92–6.87 (m, 4 H), 4.29 (q, J = 7.1 Hz, 4 H), 1.30 (t, J = 7.1 Hz, 6 H).
13C NMR (151 MHz, CDCl3): δ = 165.78, 165.69, 140.60, 130.54, 130.33, 130.26, 129.06, 128.64, 128.51, 128.47, 127.50, 61.25, 14.30.
Anal. Calcd for C33H28N2O4S: C, 72.24; H, 5.14; N, 5.11; S, 5.84. Found: C, 71.86; H, 5.15; N, 5.02; S, 5.96.
#
1,3-Bis(4-ethoxycarbonylphenyl)-4,5-diphenylimidazolium Perchlorate (3a)
Reaction of imidazolinthione 2a (13.72 g, 25 mmol) and H2O2 (8.8 mL, 35%) in acetic acid (90 mL) afforded 3a. An analytically pure sample was obtained by recrystallization from CHCl3/Et2O.
Yield: 88% (13.6 g); mp 336–338 °C (dec.).
IR (ATR): 1715s, 1607w, 1542m, 1276s, 1086s, 1020m, 772s, 702s, 621s cm–1.
1H NMR (601 MHz, DMSO-d 6): δ = 10.31 (s, 1 H), 8.14 (d, J = 8.5 Hz, 4 H), 7.70 (d, J = 8.5 Hz, 4 H), 7.41 (t, J = 7.3 Hz, 2 H), 7.36 (t, J = 7.4 Hz, 4 H), 7.26 (d, J = 7.2 Hz, 4 H), 4.35 (q, J = 7.1 Hz, 4 H), 1.34 (t, J = 7.1 Hz, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 165.05, 138.22, 137.58, 132.10, 132.06, 131.33, 131.04, 130.68, 129.35, 127.44, 125.07, 61.87, 14.54.
Anal. Calcd for C33H29ClN2O8: C, 64.24; H, 4.74; N, 4.54. Found: C, 64.05; H, 4.67; N, 4.42.
#
1,3-Bis(4-ethoxycarbonylphenyl)-4,5-diphenylimidazolium Chloride (4a)
Reaction of imidazolinthione 2a (13.72 g, 25 mmol) and H2O2 (18 mL, 35%) in acetic acid (90 mL) afforded 4a after crystallization from CH2Cl2/EtOAc.
Yield: 68% (9.4 g); mp 277–278 °C.
IR (ATR): 1702s, 1608w, 1560m, 1260s, 1175w, 1095s, 1025m, 869s, 771s, 692s cm–1.
1H NMR (601 MHz, DMSO-d 6): δ = 10.39 (s, 1 H), 8.15–8.11 (m, 4 H), 7.73 (d, J = 8.6 Hz, 4 H), 7.42–7.39 (m, 2 H), 7.36 (t, J = 7.4 Hz, 4 H), 7.30–7.26 (m, 4 H), 4.35 (q, J = 7.1 Hz, 4 H), 1.34 (t, J = 7.1 Hz, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 165.07, 138.26, 137.62, 132.06, 132.05, 131.36, 130.99, 130.65, 129.32, 127.50, 125.12, 61.86, 14.55.
Anal. Calcd for C33H29ClN2O4: C, 71.67; H, 5.29; N, 5.07. Found: C, 71.28; H, 5.20; N, 4.86.
#
N,N′-Bis(4-bromophenyl)thiourea (1b)[20] [21]
According to GP B, reaction of 4-bromoaniline (86.0 g, 0.5 mol) and carbon disulfide (45 mL, 0.75 mol), afforded 1b. An analytically pure sample was obtained by recrystallization from ethanol.
Yield: 85% (81.8 g); mp 188–189 °C (Lit.[20] 188 °C).
IR (ATR): 3206m, 3013m, 1588m, 1530s, 1482s, 1305s, 1068s, 1008s, 821s, 717s cm–1.
1H NMR (601 MHz, DMSO-d 6): δ = 9.94 (s, 2 H), 7.54–7.51 (m, 4 H), 7.49–7.45 (m, 4 H).
13C NMR (151 MHz, DMSO-d 6): δ = 180.09, 139.25, 131.74, 126.11, 117.04.
NMR spectroscopic data for 1b match those previously described.[21]
#
1,3-Bis(4-bromophenyl)-4,5-diphenylimidazolin-2-thione (2b)
Reaction of thiourea 1b (77.22 g, 0.2 mol) and benzoin (42.42 g, 0.2 mol) in acetic acid (200 mL) for 11 h afforded 2b.
Yield: 86% (96.8 g); mp 319–320 °C.
IR (ATR): 2948w, 1487s, 1356s, 1069m, 1012m, 749s, 696s, 661m cm–1.
1H NMR (601 MHz, DMSO-d 6): δ = 7.63–7.59 (m, 2 H), 7.37–7.33 (m, 2 H), 7.25–7.19 (m, 3 H), 7.16 (dt, J = 3.9, 2.3 Hz, 2 H).
13C NMR (151 MHz, DMSO-d 6): δ = 165.29, 136.55, 132.24, 131.75, 131.10, 129.10, 128.72, 128.51, 128.07, 122.03.
Anal. Calcd for C27H18Br2N2S: C, 57.67; H, 3.23; N, 4.98; S, 5.70. Found: C, 57.38; H, 3.30; N, 4.79; S, 5.92.
#
1,3-Bis(4-bromophenyl)-4,5-diphenylimidazolium Perchlorate (3b)
Reaction of imidazolinethione 2b (22.49 g, 40 mmol) and H2O2 (13.7 mL, 35%) in acetic acid (90 mL) afforded product 3b.
Yield: 90% (22.6 g); mp 319–320 °C (dec.).
IR (ATR): 1545m, 1485s, 1079s, 1012s, 1013s, 831m, 751s, 696s cm–1.
1H NMR (601 MHz, DMSO-d 6): δ = 10.18 (s, 1 H), 7.84–7.80 (m, 4 H), 7.52–7.48 (m, 4 H), 7.43–7.39 (m, 2 H), 7.39–7.34 (m, 4 H), 7.28–7.25 (m, 4 H).
13C NMR (151 MHz, DMSO-d 6): δ = 138.03, 133.30, 133.27, 132.00, 131.36, 130.64, 129.32, 128.99, 125.12, 124.33.
Anal. Calcd for C27H19Br2ClN2O4: C, 51.42; H, 3.04; N, 4.44. Found: C, 51.19; H, 3.09; N, 4.35.
#
1,3-Bis(4-bromophenyl)-4,5-diphenylimidazolium Chloride (4b)
Reaction of imidazolinethione 2b (22.49 g, 40 mmol) and H2O2 (12 mL, 35%) in acetic acid (100 mL) afforded product 4b after crystallization from CH2Cl2/EtOAc.
Yield: 73% (16.5 g); mp 292–293 °C (dec.).
IR (ATR): 2970br, 1547s, 1483s, 1249m, 1017s, 830s, 750s, 695s cm–1.
1H NMR (601 MHz, CDCl3): δ = 10.56 (s, 1 H), 7.58 (d, J = 8.7 Hz, 4 H), 7.49 (d, J = 8.7 Hz, 4 H), 7.29 (t, J = 7.5 Hz, 2 H), 7.21 (t, J = 7.9 Hz, 4 H), 7.11 (dd, J = 8.2, 1.0 Hz, 4 H).
13C NMR (151 MHz, CDCl3): δ = 137.41, 133.11, 132.32, 132.26, 130.91, 130.40, 129.04, 128.08, 124.99, 124.36.
Anal. Calcd for C27H19Br2ClN2: C, 57.22; H, 3.38; N, 4.94. Found: C, 56.84; H, 3.42; N, 4.85.
#
N,N′-Bis(4-methoxyphenyl)thiourea (1c)[22] [23] [24]
According to GP A, reaction of p-anisidine (123.2 g, 1 mol) and carbon disulfide (33 mL, 0.55 mol), afforded 1c after recrystallization from EtOH.
Yield: 78% (224.9 g); mp 194–195 °C (dec.) (Lit. 188–189 °C,[22] 198 °C[23]).
IR (ATR): 3222br, 1612m, 1537s, 1507s, 1337s, 1284s, 1235s, 1032s, 837s, 725s, 671s, 581s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.37 (s, 2 H), 7.29 (d, J = 8.9 Hz, 4 H), 6.86 (d, J = 8.9 Hz, 4 H), 3.71 (s, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 180.80, 157.07, 132.81, 126.61, 114.19, 55.78.
NMR spectroscopic data of 1c match those previously described.[24]
#
1,3-Bis-(4-methoxyphenyl)-4,5-diphenylimidazolin-2-thione (2c)
Reaction of thiourea 1c (34.41 g, 0.12 mol) and benzoin (25.2 g, 0.12 mol) in acetic acid (100 mL) for 11 h and crystallization of the resultant product from EtOH afforded 2c.
Yield: 88% (48.87 g); mp 271–272 °C.
IR (ATR): 1597m, 1511s, 1441s, 1340s, 1298s, 1241s, 1165s, 1028s, 836s, 784s, 696s, 576s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 7.23 (d, J = 8.6 Hz, 4 H), 7.13 (dt, J = 8.2, 4.1 Hz, 6 H), 7.09 (d, J = 6.6 Hz, 4 H), 6.88 (d, J = 8.6 Hz, 4 H), 3.71 (s, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 165.95, 159.23, 131.14, 130.76, 130.18, 128.81, 128.69, 128.61, 114.36, 55.82.
Anal. Calcd for C29H24N2O2S: C, 74.97; H, 5.21; N, 6.03; S, 6.90. Found: C, 74.69; H, 5.20; N, 5.95; S, 6.92.
#
1,3-Bis(4-methoxyphenyl)-4,5-diphenylimidazolium Perchlorate (3c)
Reaction of imidazolinethione 2c (10.0 g, 22 mmol) and H2O2 (7.8 mL, 35%) in acetic acid (100 mL) afforded 3c after recrystallization from MeOH (1 L).
Yield: 74% (8.45 g); mp 289–291 °C.
IR (ATR): 1546m, 1508s, 1447m, 1253s, 1177m,1088s, 833s, 697s, 621s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.97 (s, 1 H), 7.48–7.40 (m, 4 H), 7.32 (dd, J = 11.1, 3.9 Hz, 2 H), 7.28 (t, J = 7.5 Hz, 4 H), 7.25–7.20 (m, 4 H), 7.04 (dd, J = 14.0, 5.5 Hz, 4 H), 3.75 (s, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 160.88, 137.83, 132.27, 131.46, 130.37, 129.13, 128.44, 126.82, 125.76, 115.28, 56.18.
Anal. Calcd for C29H25ClN2O6: C, 65.35; H, 4.73; N, 5.26. Found: C, 64.64; H, 4.52; N, 4.95.
#
1,3-Bis(4-methoxyphenyl)-4,5-diphenylimidazolium Chloride (4c)[8e]
Reaction of imidazolinethione 2c (23.2 g, 50 mmol) and H2O2 (18 mL, 35%) in acetic acid (150 mL) afforded 4c. An analytical pure sample was obtained by recrystallization from CH2Cl2/Et2O. Yield: 89% (20.83 g); mp 143–144 °C.
IR (ATR): 3231w, 1550s, 1507s, 1449m, 1236s, 1177s, 1019s, 834s, 785s, 699s cm–1.
1H NMR (601 MHz, CDCl3): δ = 10.37 (s, 1 H), 7.63 (d, J = 8.9 Hz, 4 H), 7.32 (t, J = 7.5 Hz, 2 H), 7.25 (t, J = 7.7 Hz, 4 H), 7.17 (d, J = 7.4 Hz, 4 H), 6.90 (d, J = 8.9 Hz, 4 H), 3.79 (s, 6 H).
1H NMR data were in accordance with reported data.[8e]
13C NMR (151 MHz, CDCl3): δ = 160.72, 137.32, 132.10, 130.95, 129.96, 128.80, 127.81, 126.11, 125.03, 114.89, 55.58.
Anal. Calcd for C29H25ClN2O2: C, 74.27; H, 5.37; N, 5.97. Found: C, 73.59; H, 5.40; N, 5.57.
#
N,N′-Bis(4-dimethylaminophenyl)thiourea (1d)[22]
According to GP A, reaction of N,N-dimethyl-p-phenylenediamine (50.0 g, 0.37 mol) and carbon disulfide (12 mL, 0.2 mol), followed by recrystallization from EtOH afforded 1d.
Yield: 72% (41.9 g); mp 189–190 °C (dec.) (Lit.[22] 185–186 °C).
IR (ATR): 3300–3100br, 2884m, 1611s, 1519s, 1367s, 1231s, 1174s, 945s, 818s, 721s, 673s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.08 (s, 2 H), 7.16 (d, J = 8.9 Hz, 4 H), 6.65 (d, J = 9.0 Hz, 4 H), 2.84 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 180.51, 148.67, 129.09, 126.39, 112.81, 40.93.
Anal. Calcd for C17H22N4S: C, 64.93; H, 7.05; N, 17.82; S, 10.20. Found: C, 64.70; H, 7.02; N, 17.71; S, 10.31.
#
1,3-Bis(4-dimethylaminophenyl)-4,5-di(4-methoxyphenyl)imidazolin-2-thione (2d)
Reaction of thiourea 1d (31.45 g, 0.1 mol) and anisoin (27.2 g, 0.1 mol) in acetic acid (100 mL) for 7 h afforded 2d after recrystallization from EtOH. An analytically pure sample was obtained by recrystallization from CHCl3/EtOH.
Yield: 61% (33.6 g); mp 250–251 °C (dec.).
IR (ATR): 1610s, 1516s, 1342s, 1247s, 1172s, 1026s, 803s, 757s cm–1.
1H NMR (601 MHz, CDCl3): δ = 7.17 (d, J = 8.2 Hz, 4 H), 6.91 (d, J = 8.4 Hz, 4 H), 6.66 (d, J = 8.4 Hz, 4 H), 6.64 (d, J = 8.4 Hz, 4 H), 3.71 (s, 6 H), 2.95 (s, 12 H).
13C NMR (151 MHz, CDCl3): δ = 165.44, 158.95, 149.83, 131.77, 129.45, 128.03, 126.22, 121.11, 113.57, 112.16, 55.06, 40.48.
Anal. Calcd for C33H34N4O2S: C, 71.97; H, 6.22; N, 10.17; S, 5.82. Found: C, 71.64; H, 6.11; N, 10.02; S, 5.39.
#
1,3-Bis(4-dimethylaminophenyl)-4,5-di(4-methoxyphenyl)imidazolium Perchlorate (3d)
Reaction of imidazolinethione 2d (10.4 g, 20 mmol) and H2O2 (6 mL, 35%) in acetic acid (90 mL) afforded, after crystallization from methanol, product 3d. An analytically pure sample was obtained by recrystallization from CHCl3/MeOH.
Yield: 31% (3.4 g); mp 174–175 °C.
IR (ATR): 1612m, 1518s, 1244s, 1180m, 1089s, 818s, 620s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.73 (s, 1 H), 7.24 (d, J = 8.6 Hz, 4 H), 7.13 (d, J = 8.4 Hz, 4 H), 6.84 (d, J = 8.5 Hz, 4 H), 6.71 (d, J = 8.8 Hz, 4 H), 3.67 (s, 6 H), 2.90 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 160.43, 151.36, 136.90, 132.88, 131.92, 127.53, 122.40, 118.13, 114.59, 112.27, 55.67, 40.34.
Anal. Calcd for C33H35ClN4O6: C, 64.02; H, 5.70; N, 9.05. Found: C, 63.62; H, 5.32; N, 8.91.
#
1,3-Bis(4-dimethylaminophenyl)-4,5-di(4-methoxyphenyl)imidazolium Chloride (4d)
Reaction of imidazolinethione 2d (12.21 g, 22 mmol) and H2O2 (8 mL, 35%) in acetic acid (80 mL) afforded 4d.
Yield: 45% (5.53 g); mp 110–112 °C.
IR (ATR): 1715m, 1518s, 1445m, 1228s, 1178s, 1025m, 821s, 605s cm–1.
1H NMR (601 MHz, CDCl3): δ = 8.52 (s, 1 H), 7.32 (d, J = 9.0 Hz, 1 H), 7.12 (d, J = 8.8 Hz, 1 H), 6.69 (d, J = 8.8 Hz, 1 H), 6.59 (d, J = 9.0 Hz, 1 H), 3.70 (s, 6 H), 2.93 (s, 12 H).
13C NMR (151 MHz, CDCl3): δ = 160.26, 150.99, 134.22, 132.55, 132.41, 127.19, 122.05, 117.56, 114.05, 112.10, 55.17, 40.24.
Anal. Calcd for C33H35ClN4O2: C, 71.40; H, 6.36; N, 10.09. Found: C, 71.01; H, 6.34; N, 9.87.
#
N,N′-Bis(3,5-dimethylphenyl)thiourea (1e)[25]
According to GP A, reaction of 3,5-dimethylaniline (121.2 g, 1 mol) and carbon disulfide (42 g, 0.55 mol), after recrystallization from EtOH (800 mL) afforded 1e.
Yield: 68% (96.1 g); mp 150–151 °C.
IR (ATR): 3347s, 3200–2900br, 1607s, 1536s, 1508s, 1303s, 1272s, 1228s, 854s, 700s, 652s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.49 (s, 2 H), 7.02 (s, 4 H), 6.73 (s, 2 H), 2.21 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 179.98, 139.74, 137.96, 126.58, 122.07, 21.48.
NMR data were in accordance with those described.[25]
#
1,3-Bis(3,5-dimethylphenyl)-4,5-diphenylimidazolin-2-thione (2e)
Reaction of thiourea 1e (67.5 g, 0.25 mol) and benzoin (53.0 g, 0.25 mol) in acetic acid (100 mL) for 10 h afforded 2e.
Yield: 87% (100.2 g); mp 239–240 °C.
IR (ATR): 1594m, 1328s, 1258w, 723s, 689s, 610s, 581s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 7.24–7.01 (m, 10 H), 6.92 (s, 6 H), 2.17 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 165.50, 138.29, 137.31, 131.08, 130.24, 128.81, 128.64, 128.58, 128.53, 127.20, 21.18.
Anal. Calcd for C31H28N2S: C, 80.83; H, 6.13; N, 6.08; S, 6.96. Found: C, 79.95; H, 6.11; N, 5.95; S, 7.32.
#
1,3-Bis(3,5-dimethylphenyl)-4,5-diphenylimidazolium Perchlorate (3e)
Reaction of imidazolinethione 2e (37.1 g, 80 mmol) and H2O2 (28 mL, 35%) in acetic acid (150 mL) afforded 3e. An analytically pure sample was obtained by recrystallization from CHCl3/Et2O.
Yield: 64% (27.1 g); mp 276–277 °C.
IR (ATR): 1542m, 1083s, 864m, 772m, 697s, 621s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 10.00 (s, 1 H), 7.36–7.32 (m, 2 H), 7.30 (t, J = 7.4 Hz, 4 H), 7.22 (d, J = 7.3 Hz, 4 H), 7.16 (s, 2 H), 7.12 (s, 4 H), 2.23 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 139.68, 137.65, 133.99, 132.19, 132.04, 131.42, 130.49, 129.18, 125.61, 124.42, 21.18.
Anal. Calcd for C31H29ClN2O4: C, 70.38; H, 5.53; N, 5.30. Found: C, 69.91; H, 5.51; N, 5.01.
#
1,3-Bis(3,5-dimethylphenyl)-4,5-diphenylimidazolium Chloride (4e)
Reaction of imidazolinthione 2e (6.9 g, 15 mmol) and H2O2 (5.3 mL, 35%) in acetic acid (50 mL) afforded 4e.
Yield: 54% (3.77 g); mp 222–224 °C (dec.).
IR (ATR): 1719m, 1540m, 1256m, 1022m, 859m, 690s cm–1.
1H NMR NMR (400 MHz, CDCl3): δ = 10.35 (s, 1 H), 7.35–7.30 (m, 2 H), 7.27 (d, J = 2.5 Hz, 2 H), 7.25–7.22 (m, 6 H), 7.19–7.14 (m, 4 H), 7.04 (s, 2 H), 2.28 (s, 12 H).
13C NMR (101 MHz, CDCl3): δ = 139.80, 137.06, 133.20, 132.00, 131.93, 130.90, 129.90, 128.70, 125.12, 123.93, 21.14.
Anal. Calcd for C31H29ClN2: C, 80.07; H, 6.29; N, 6.02. Found: C, 79.52; H, 6.22; N, 5.85.
#
1,3-Bis(3,5-dimethylphenyl)-4,5-di(4-methoxyphenyl)imidazolin-2-thione (2f)
Reaction of thiourea 1e (96.0 g, 0.34 mol) and anisoin (91.0 g, 0.34 mol) in acetic acid (400 mL) for 12 h, after recrystallization from EtOH (1 L) afforded 2f.
Yield: 91% (161.2 g); mp 267–268 °C (dec.).
IR (ATR): 1605m, 1505s, 1364s, 1327s, 1293s, 1246s, 1174s, 827s, 755s cm–1.
1H NMR (400 MHz, CDCl3): δ = 6.97–6.93 (m, 6 H), 6.92–6.87 (m, 4 H), 6.65–6.60 (m, 4 H), 3.69 (s, 6 H), 2.27 (s, 12 H).
13C NMR (101 MHz, CDCl3): δ = 164.71, 159.14, 138.47, 137.00, 131.72, 130.37, 128.04, 126.74, 120.70, 113.57, 55.08, 21.23.
Anal. Calcd for C33H32N2O2S: C, 76.12; H, 6.19; N, 5.38; S, 6.16. Found: C, 75.89; H, 6.20; N, 5.19; S, 6.49.
#
1,3-Bis(3,5-dimethylphenyl)-4,5-di(4-methoxyphenyl)imidazolium Perchlorate (3f)
Reaction of imidazolinethione 2f (52.0 g, 0.1 mol) and H2O2 (34 mL, 35%) in acetic acid (150 mL) afforded 3f.
Yield: 89% (52.3 g); mp 274–275 °C (dec.).
IR (ATR): 1614m, 1506m, 1473m, 1255s, 1082s, 841s, 620s cm–1.
1H NMR (400 MHz, CDCl3): δ = 9.70 (s, 1 H), 7.22 (s, 4 H), 7.09 (t, J = 8.4 Hz, 4 H), 7.02 (s, 2 H), 6.72 (d, J = 8.4 Hz, 4 H), 3.72 (s, 6 H), 2.27 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 160.63, 139.62, 137.09, 134.17, 132.89, 132.12, 131.84, 124.52, 117.63, 114.63, 55.71, 21.20.
Anal. Calcd for C33H33ClN2O6: C, 67.28; H, 5.65; N, 4.76. Found: C, 66.57; H, 5.61; N, 4.65.
#
1,3-Bis(3,5-dimethylphenyl)-4,5-di(4-methoxyphenyl)imidazolium Chloride (4f)
Reaction of imidazolinethione 2f (104.0 g, 0.2 mol) and H2O2 (53 mL, 35%) in acetic acid (200 mL) afforded 4f.
Yield: 81% (84.5 g); mp 216–217 °C (dec.).
IR (ATR): 1543m, 1249s, 1176s, 1023s, 833s, 695s, 584s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.94 (s, 1 H), 7.18–7.13 (m, 10 H), 6.84 (d, J = 7.9 Hz, 4 H), 3.66 (d, J = 0.7 Hz, 6 H), 2.23 (s, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 160.65, 139.57, 137.14, 134.19, 132.93, 132.08, 131.85, 124.55, 117.68, 114.62, 55.74, 21.18.
Anal. Calcd for C33H33ClN2O2: C, 75.48; H, 6.33; Cl, N, 5.34. Found: C, 74.65; H, 6.24; N, 5.06.
#
N,N′-Bis(2-methoxyphenyl)thiourea (1g)[26]
According to GP A, reaction of o-anisidine (61.53 g, 0.5 mol) and carbon disulfide (22.8 g, 0.3 mol), after recrystallization from EtOH (200 mL) afforded 1g.
Yield: 56% (39.4 g); mp 136–137 °C (Lit.[26] 132–134 °C).
IR (ATR): 3200–3100 br, 2958 br, 1509s, 1490s, 1455s, 1313s, 1258s, 1231s, 1163s, 1112s, 1024s, 778s, 754s, 645s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.33 (s, 1 H), 7.93 (dd, J = 7.9, 1.0 Hz, 1 H), 7.14–7.09 (m, 1 H), 7.03 (dd, J = 8.2, 1.1 Hz, 1 H), 6.92–6.87 (m, 1 H), 3.79 (s, 3 H).
13C NMR (151 MHz, DMSO-d 6): δ = 180.00, 152.26, 128.15, 126.31, 120.34, 111.96, 56.23.
Anal. Calcd for C15H16N2O2S: C, 62.48; H, 5.59; N, 9.71; S, 11.12. Found: C, 62.41; H, 5.60; N, 9.65; S, 11.10.
#
1,3-Bis(2-methoxyphenyl)-4,5-diphenylimidazolin-2-thione (2g)
Reaction of thiourea 1g (39.4 g, 0.14 mol) and benzoin (28.8 g, 0.14 mol) in acetic acid (70 mL) for 6 h afforded 2g. An analytically pure sample was obtained by recrystallization from CHCl3.
Yield: 61% (38.5 g); mp 242–243 °C.
IR (ATR): 1502m, 1377m, 1346s, 1253m, 1019m, 750s, 700s, 570s cm–1.
1H NMR (601 MHz, CDCl3): δ = 7.59 (d, J = 7.6 Hz, 2 H), 7.32 (t, J = 7.7 Hz, 2 H), 7.14–7.10 (m, 2 H), 7.06 (dd, J = 15.0, 7.6 Hz, 6 H), 7.02 (d, J = 7.4 Hz, 4 H), 6.83 (d, J = 8.3 Hz, 2 H), 3.53 (s, 6 H).
13C NMR (151 MHz, CDCl3): δ = 154.73, 131.27, 130.44, 129.91, 128.77, 128.71, 127.89, 127.74, 125.95, 120.66, 112.35, 55.54.
Anal. Calcd for C29H24N2O2S: C, 74.97; H, 5.21; N, 6.03; S, 6.90. Found: C, 74.37; H, 5.20; N, 5.89; S, 6.91.
#
1,3-Bi-(2-methoxyphenyl)-4,5-diphenylimidazolium Perchlorate (3g)
Reaction of imidazolinethione 2g (23.1 g, 50 mmol) and H2O2 (18 mL, 35%) in acetic acid (50 mL) afforded 3g.
Yield: 54% (14.4 g); mp 197–200 °C.
IR (ATR): 1548m, 1442m, 1259m, 1083s, 761s, 695s, 623s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.95 (s, 1 H), 7.56 (dd, J = 7.8, 1.4 Hz, 2 H), 7.50–7.43 (m, 2 H), 7.29–7.23 (m, 2 H), 7.20 (t, J = 7.6 Hz, 4 H), 7.12 (dd, J = 12.1, 4.6 Hz, 2 H), 7.11–7.07 (m, 4 H), 7.04 (dd, J = 11.3, 4.0 Hz, 2 H), 3.56 (s, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 153.89, 139.22, 133.12, 132.37, 130.56, 130.36, 129.08, 128.98, 125.65, 122.30, 121.50, 113.64, 56.58.
Anal. Calcd for C29H25ClN2O6: C, 65.35; H, 4.73; N, 5.26. Found: C, 64.84; H, 4.71; N, 5.23.
#
1,3-Bis(2-methoxyphenyl)-4,5-diphenylimidazolium Chloride (4g)
Reaction of imidazolinethione 2g (18.57 g, 40 mmol) and H2O2 (14 mL, 35%) in acetic acid (100 mL) afforded 4g.
Yield: 58% (10.9 g); mp 181–183 °C.
IR (ATR): 1547m, 1286s, 1256s, 1127w, 1018s, 752s, 701s, 666m cm–1.
1H NMR (601 MHz, CDCl3): δ = 8.76 (s, 1 H), 7.72 (dd, J = 7.9, 1.4 Hz, 2 H), 7.43 (td, J = 8.4, 1.5 Hz, 2 H), 7.26 (dq, J = 3.4, 1.6 Hz, 2 H), 7.21–7.15 (m, 8 H), 7.01 (td, J = 7.8, 0.8 Hz, 2 H), 6.96 (d, J = 8.4 Hz, 2 H), 3.70 (s, 6 H).
13C NMR (151 MHz, CDCl3): δ = 153.46, 137.02, 132.68, 132.40, 130.42, 129.71, 129.27, 128.43, 125.34, 122.00, 121.51, 112.07, 55.90.
Anal. Calcd for C29H25ClN2O2: C, 74.27; H, 5.37; N, 5.97. Found: C, 73.71; H, 5.25; N, 5.78.
#
N,N′-Bis(2,4-dimethylphenyl)thiourea (1h)[9b]
According to GP A, reaction of 2,4-dimethylaniline (65.0 g, 0.5 mol) and carbon disulfide (30 mL, 0.5 mol), after recrystallization from EtOH (300 mL) afforded 1h.
Yield: 98% (70.1 g); mp 155–156 °C (Lit.[9b] 214 °C).
IR (ATR): 3196br, 1550s, 1462s, 1263s, 1144s, 696s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 8.91 (s, 2 H), 7.07 (d, J = 7.9 Hz, 2 H), 7.01 (s, 2 H), 6.95 (d, J = 7.8 Hz, 2 H), 2.23 (s, 6 H), 2.16 (s, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ = 181.91, 136.21, 135.77, 135.39, 131.40, 128.66, 127.22, 21.11, 18.24.
Anal. Calcd for C17H20N2S: C, 71.79; H, 7.09; N, 9.85; S, 11.27. Found: C, 71.15; H, 7.05; N, 9.65; S, 11.92.
#
1,3-Bis(2,4-dimethylphenyl)-4,5-diphenylimidazolin-2-thione (2h)
Reaction of thiourea 1h (20.2 g, 70 mmol) and benzoin (14.85 g, 70 mmol) in acetic acid (100 mL) for 7 h afforded 5h, as a mixture of two isomers (4:3) after recrystallization from CHCl3/EtOH in isomer ratio 7:2.
Yield: 63% (20.3 g); mp 235–237 °C.
IR (ATR): 1502m, 1339s, 757m, 694s, 576s cm–1.
1H (601 MHz, CDCl3): δ (major isomer) = 7.14 (d, J = 7.9 Hz, 2 H), 7.10–7.05 (m, 2 H), 7.01 (dd, J = 10.3, 4.7 Hz, 4 H), 6.98 (d, J = 7.8 Hz, 2 H), 6.96–6.91 (m, 6 H), 2.23 (s, 6 H), 2.07 (s, 6 H).
13C NMR (101 MHz, CDCl3): δ = 165.17, 139.17, 135.96, 133.53, 131.88, 130.17, 129.41, 129.04, 128.18, 128.16, 127.56, 21.28, 18.16.
Anal. Calcd for C31H28N2S: C, 80.83; H, 6.13; N, 6.08; S, 6.96. Found: C, 80.54; H, 6.15; N, 5.96; S, 6.95.
#
1,3-Bis(2,4-dimethylphenyl)-4,5-diphenylimidazolium Perchlorate (3h)
Reaction of imidazolinethione 2h (9.12 g, 20 mmol) and H2O2 (7 mL, 35%) in acetic acid (50 mL) afforded 3h.
Yield: 51% (5.4 g); mp 262–264 °C.
IR (ATR): 1537m, 1079s, 822m, 784m, 622m cm–1.
1H NMR (600 MHz, 80 °C, DMSO-d 6): δ = 9.84 (s, 1 H), 7.50 (d, J = 7.6 Hz, 2 H), 7.31 (ddd, J = 6.2, 3.2, 1.5 Hz, 2 H), 7.28–7.22 (m, 8 H), 7.21–7.15 (m, 4 H), 2.30 (s, 6 H), 2.13 (s, 6 H).
13C NMR (151 MHz, 80 °C, DMSO-d 6): δ = 141.49, 138.43, 134.85, 132.95, 132.30, 131.11, 130.67, 130.52, 129.06, 128.59, 128.16, 125.54, 21.10, 17.42.
Anal. Calcd for C31H29ClN2O4: C, 70.38; H, 5.53; N, 5.30. Found: C, 70.32; H, 5.51; N, 5.25.
#
1,3-Bis(2,4-dimethylphenyl)-4,5-diphenylimidazolium Chloride (4h)
Reaction of imidazolinethione 2h (4.61 g, 10 mmol) and H2O2 (3.5 mL, 35%) in acetic acid (25 mL) afforded 4h.
Yield: 69% (3.2 g); mp 248–249 °C (dec.).
IR (ATR): 2892m, 1531s, 1443m, 1228s, 825m, 785s, 696s, 565m cm–1.
1H NMR (600 MHz, 80 °C, DMSO-d 6): δ = 9.96 (s, 1 H), 7.54 (d, J = 7.8 Hz, 2 H), 7.33–7.28 (m, 2 H), 7.27–7.21 (m, 8 H), 7.18 (s, 2 H), 7.16 (d, J = 8.0 Hz, 2 H), 2.29 (s, 6 H), 2.14 (s, 6 H).
13C NMR (151 MHz, 80 °C, DMSO-d 6): δ = 141.40, 138.52, 134.84, 132.89, 132.24, 131.13, 130.71, 130.47, 129.02, 128.67, 128.10, 125.60, 21.10, 17.46.
Anal. Calcd for C31H29ClN2: C, 80.07; H, 6.29; N, 6.02. Found: C, 79.66; H, 6.22; N, 5.95.
#
N,N′-Bis(2-methyl-5-tert-butylphenyl)thiourea (1i)
According to GP A, reaction of 2-methyl-5-tert-butylaniline (194.0 g, 92%, 1.1 mol) and carbon disulfide (79 g, 1 mol), after recrystallization from 90% EtOH (600 mL) afforded 1i.
Yield: 62% (125.1 g); mp 152–153 °C.
IR (ATR): 3187br, 2962s, 1535s, 1410s, 1293s, 1258s, 1222s, 808s, 733s, 600s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.02 (s, 2 H), 7.26 (s, 2 H), 7.15 (d, J = 8.0 Hz, 2 H), 7.12 (d, J = 8.0 Hz, 2 H), 2.19 (s, 6 H), 1.23 (s, 18 H).
13C NMR (151 MHz, DMSO-d 6): δ = 181.43, 149.10, 137.84, 132.14, 130.49, 125.20, 123.84, 34.59, 31.66, 17.88.
Anal. Calcd for C23H32N2S: C, 74.95; H, 8.75; N, 7.60; S, 8.70. Found: C, 74.25; H, 8.65; N, 7.41; S, 9.25.
#
1,3-Bis(2-methyl-5-tert-butylphenyl)-4,5-di(4-methoxyphenyl)-imidazolin-2-thione (2i)
Reaction of thiourea 1i (29.5 g, 80 mmol) and anisoin (21.8 g, 80 mmol) in acetic acid (400 mL) for 22 h, followed by purification by column chromatography using hexane–EtOAc (5:1) afforded 2i as a mixture of two isomers (1:1). Recrystallization from diethyl ether/hexane gave the pure isomer.
Yield: 76% (36.8 g); mp 207–208 °C.
IR (ATR): 3000–2900br, 1505s, 1346s, 1252s, 1022m, 830s, 626m, 562s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 7.22 (dd, J = 8.0, 1.6 Hz, 2 H), 7.18 (s, 2 H), 7.12 (d, J = 8.0 Hz, 2 H), 7.00 (d, J = 8.7 Hz, 4 H), 6.63 (d, J = 8.7 Hz, 4 H), 3.58 (s, 6 H), 2.10 (s, 6 H), 1.18 (s, 18 H).
13C NMR (151 MHz, DMSO-d 6): δ = 163.58, 159.52, 149.46, 136.29, 133.66, 132.31, 130.66, 128.25, 127.28, 125.87, 120.91, 113.92, 55.54, 34.59, 31.50, 17.99.
Anal. Calcd for C39H44N2O2S: C, 77.45; H, 7.33; N, 4.63; S, 5.30. Found: C, 77.05; H, 7.35; N, 4.45; S, 5.30.
#
1,3-Bis(2-methyl-5-tert-butylphenyl)-4,5-di(4-methoxyphenyl)imidazolium perchlorate (3i)
H2O2 (52 mL, 35%) was carefully added to the reaction solution of imidazolinethione 2i (150 mmol) in acetic acid (250 mL) from the previous step. The temperature was allowed to rise to 60 °C and the mixture was stirred at ambient temperature for 4 h. All volatiles were removed by rotary evaporation and the residue was dissolved in MeOH (200 mL) and treated with a solution of NaClO4 (28.1 g, 0.2 mol) dissolved in a 2:1 (v/v) mixture of methanol/water (200 mL). A white solid precipitated. The suspension was further stirred in an ice bath and the precipitate was then filtered off and washed with water, diethyl ether and dried in vacuo to obtain product 3i.
Yield: 70% (71.1 g); mp 264–265 °C.
IR (ATR): 1521w, 1253m, 1089s, 1027m, 837m, 621m cm–1.
1H NMR (601 MHz, CDCl3): δ = 8.60 (s, 1 H), 7.87 (s, 2 H), 7.39 (dd, J = 8.0, 1.5 Hz, 2 H), 7.17 (d, J = 8.1 Hz, 2 H), 7.09 (d, J = 8.1 Hz, 4 H), 6.72–6.67 (m, 4 H), 3.71 (s, 6 H), 2.11 (s, 6 H), 1.28 (s, 18 H).
13C NMR (151 MHz, CDCl3): δ = 160.51, 151.77, 134.76, 132.76, 132.32, 132.08, 130.61, 127.77, 126.65, 117.11, 114.13, 55.23, 34.77, 31.06, 17.02.
Anal. Calcd for C39H45ClN2O6: C, 69.58; H, 6.74; N, 4.16. Found: C, 69.23; H, 6.56; N, 4.05.
#
1,3-Bis(2-methyl-5-tert-butylphenyl)-4,5-di(4-methoxyphenyl)-imidazolium Chloride Methanol (4i)
Reaction of imidazolinethione 2i (36.29 g, 60 mmol) and H2O2 (18 mL, 35%) in acetic acid (150 mL) afforded 4i. An analytically pure sample was obtained by recrystallization from MeOH/Et2O at –18 °C.
Yield: 70% (26.6 g); mp 128–130 °C.
IR (ATR): 2963br, 1505s, 1444m, 1247s, 1182s, 1033s, 832s, 655m cm–1.
1H NMR (601 MHz, CDCl3): δ = 10.69 (s, 1 H), 7.51 (br. s., 2 H), 7.37 (dd, J = 8.1, 1.9 Hz, 2 H), 7.21 (d, J = 8.1 Hz, 2 H), 6.96 (d, J = 8.1 Hz, 4 H), 6.74–6.67 (m, 4 H), 3.73 (s, 6 H), 3.41(s, 3 H), 2.23 (s, 6 H), 1.25 (s, 18 H).
13C NMR (101 MHz, CDCl3): δ = 160.51, 150.86, 138.17, 132.22, 131.74, 131.66, 131.20, 127.73, 125.65, 117.26, 114.26, 55.29, 50.54 (MeOH), 34.59, 31.11, 17.50.
Anal. Calcd for C41H48ClN2O4: C, 74.92; H, 7.70; N, 4.37. Found: C, 74.56; H, 7.71; N, 4.35.
#
N,N′-Bis-(2-isopropylphenyl)thiourea (1j)
According to GP B, reaction of 2-isopropylaniline (135.0 g, 1.0 mol) and carbon disulfide (42 g, 0.55 mol) in diglyme (200 mL), after recrystallization from MeOH (800 mL) afforded 1j.
Yield: 56% (100.3 g); mp 157–158 °C.
IR (ATR): 3250–3050br, 1531s, 1498s, 1258s, 767m, 573s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 9.02 (s, 2 H), 7.29 (d, J = 7.7 Hz, 2 H), 7.24–7.20 (m, 2 H), 7.18–7.13 (m, 4 H), 3.14 (hept, J = 6.9 Hz, 2 H), 1.15 (d, J = 6.9 Hz, 12 H).
13C NMR (151 MHz, DMSO-d 6): δ = 182.89, 146.08, 137.02, 129.77, 127.79, 126.40, 126.22, 28.15, 23.70.
Anal. Calcd for C19H24N2S: C, 73.03; H, 7.74; N, 8.97; S, 10.26. Found: C, 72.54; H, 7.61; N, 8.65; S, 10.65.
#
1,3-Bis-(2-isopropylphenyl)-4,5-diphenylimidazolin-2-thione (2j)
Reaction of thiourea 1j (9.77 g, 20 mmol) and benzoin (16.97 g, 80 mmol) in acetic acid (50 mL) for 17 h afforded 2j as a mixture of two isomers (2:1).
Yield: 31% (12.1 g).
1H NMR (600 MHz, DMSO-d 6): δ (major isomer) = 7.37–7.30 (m, 6 H), 7.23–7.18 (m, 2 H), 7.16–7.07 (m, 10 H), 2.79 (hept, J = 6.8 Hz, 2 H), 1.19 (d, J = 6.8 Hz, 6 H), 0.98 (d, J = 6.9 Hz, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ (major isomer) = 166.16, 146.73, 134.97, 131.21, 130.65, 130.08, 129.05, 128.94, 128.51, 128.25, 127.14, 126.67, 28.56, 24.37, 23.20.
Filtration of the component that was insoluble in hot EtOH gave the pure minor isomer. Mp 250–251 °C.
IR (ATR): 2962w, 1490m, 1340s, 751s, 695s, 598s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ (minor isomer) = 7.50–7.47 (m, 1 H), 7.26 (dd, J = 7.3, 1.4 Hz, 1 H), 7.23–7.18 (m, 4 H), 7.01 (dd, J = 4.8, 4.3 Hz, 2 H), 2.62–2.54 (m, 1 H), 1.11 (d, J = 6.8 Hz, 3 H), 0.61 (d, J = 6.8 Hz, 3 H).
13C NMR (151 MHz, DMSO-d 6): δ (minor isomer) = 166.65, 146.52, 134.95, 131.40, 130.98, 130.13, 128.90, 128.61, 128.58, 128.35, 127.09, 126.71, 28.35, 24.75, 22.55.
Anal. Calcd for C33H32N2S: C, 81.11; H, 6.60; N, 5.73; S, 6.56. Found: C, 79.88; H, 6.65; N, 5.54; S, 7.01.
#
1,3-Bis(2-isopropylphenyl)-4,5-diphenylimidazolium Perchlorate (3j)
Reaction of imidazolinethione 2j (12.2 g, 25 mmol) and H2O2 (9 mL, 35%) in acetic acid (100 mL) afforded 3j as a mixture of two isomers (2:1).
Yield: 81% (11.26 g); mp 238–241 °C.
IR (ATR): 1547m, 1445m, 1090s, 763s, 703s, 622s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ (major isomer) = 10.16 (s, 1 H), 7.69–7.67 (m, 2 H), 7.58–7.50 (m, 2 H), 7.33–7.20 (m, 14 H), 2.77 (sept, J = 6.8 Hz, 2 H), 1.13 (d, J = 6.8 Hz, 6 H), 1.00 (d, J = 6.8 Hz, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ (major isomer) = 145.59, 138.57, 133.07, 132.21, 131.41, 131.34, 130.59, 129.00, 128.86, 127.69, 127.57, 125.22, 28.35, 25.17, 22.62.
1H NMR (600 MHz, DMSO-d 6): δ (minor isomer) = 10.09 (s, 1 H), 7.88–7.85 (m, 2 H), 7.58–7.49 (m, 4 H), 7.49–7.44 (m, 4 H), 7.41–7.37 (m, 4 H), 7.33–7.20 (m, 4 H), 2.74 (sept, J = 6.8 Hz, 2 H), 1.12 (d, J = 6.8 Hz, 6 H), 0.66 (d, J = 6.8 Hz, 6 H).
13C NMR (151 MHz, DMSO-d 6): δ (minor isomer) = 145.83, 138.18, 132.97, 132.27, 131.45, 131.32, 130.50, 129.43, 129.01, 127.80, 127.50, 125.50, 27.92, 25.39, 22.42.
Anal. Calcd for C33H33ClN2O4: C, 70.38; H, 5.53; N, 5.30. Found: C, 70.35; H, 5.69; N, 5.12.
#
N,N′-Bis(2-tert-butylphenyl)thiourea (1k)
According to GP B, reaction of 2-tert-butylaniline (25.0 g, 0.168 mol) and carbon disulfide (7 g, 90 mmol) in diglyme (100 mL), after recrystallization from EtOH (80 mL) afforded 1k.
Yield: 38% (10.9 g); mp 165–166 °C.
IR (ATR): 3374m, 3218br, 2961br, 1526s, 1479s, 1256s, 753s, 632s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 8.91 (s, 2 H), 7.40–7.35 (m, 2 H), 7.19–7.15 (m, 6 H), 1.36 (s, 18 H).
13C NMR (151 MHz, DMSO-d 6): δ = 183.20, 146.97, 138.03, 133.20, 127.60, 127.23, 126.70, 35.33, 31.38.
Anal. Calcd for C21H28N2S: C, 74.07; H, 8.29; N, 8.23; S, 9.41. Found: C, 73.85; H, 8.26; N, 8.20; S, 9.48.
#
2-[(2-tert-Butylphenyl)amino]-1,2-diphenylethanone (5)
Reaction of thiourea 1k (11.9 g, 35 mmol) and benzoin (7.41 g, 35 mmol) in acetic acid (50 mL) for 17 h, after recrystallization from diethyl ether/hexane afforded 5.
Yield: 51% (6.1 g); mp 154–155 °C.
IR (ATR): 3461m, 2956br, 1683s, 1506s, 1448s, 1250s, 744s, 693s, 635s, 595s cm–1.
1H NMR (600 MHz, DMSO-d 6): δ = 8.14 (d, J = 8.0 Hz, 2 H), 7.61–7.54 (m, 1 H), 7.50 (d, J = 8.0 Hz, 2 H), 7.47 (t, J = 7.6 Hz, 2 H), 7.23 (t, J = 7.6 Hz, 2 H), 7.15–7.07 (m, 2 H), 6.89 (t, J = 7.6 Hz, 1 H), 6.72 (d, J = 8.1 Hz, 1 H), 6.53–6.47 (m, 2 H), 5.87 (d, J = 5.2 Hz, 1 H), 1.45 (s, 9 H).
13C NMR (151 MHz, DMSO-d 6): δ = 198.00, 155.50, 148.73, 143.79, 138.82, 134.86, 134.35, 133.25, 129.59, 129.30, 129.29, 128.69, 128.24, 127.15, 126.41, 117.17, 113.05, 61.88, 34.32, 30.17.
Anal. Calcd for C24H25NO: C, 83.93; H, 7.34; N, 4.08. Found: C, 83.65; H, 7.29; N, 4.01.
#
N , N ′-Bis(3-pyridinyl)thiourea (1l)[27] [28]
According to GP B, reaction of 3-aminopyridine (94.1 g, 1 mol) and carbon disulfide (38 mL, 0.63 mol) in diglyme (250 mL), after recrystallization from EtOH/H2O (200/50 mL) afforded 1l. An analytically pure sample was obtained by recrystallization from ethanol.
Yield: 35% (40.6 g); mp 178–179 °C (Lit.[27] 178–180 °C).
IR (ATR): 2971m, 2902m, 1580s, 1536s, 1414s, 1268s, 1252s, 1047s, 1024s, 1016s, 753s, 717s, 701s cm–1.
1H NMR (601 MHz, DMSO-d 6): δ = 10.08 (s, 2 H), 8.63 (dd, J = 2.6, 0.6 Hz, 2 H), 8.37 (dd, J = 4.7, 1.5 Hz, 2 H), 7.97 (ddd, J = 8.2, 2.6, 1.5 Hz, 2 H), 7.40 (ddd, J = 8.2, 4.7, 0.6 Hz, 2 H).
13C NMR (151 MHz, DMSO-d 6): δ = 181.47, 146.04, 145.99, 136.56, 132.04, 123.73.
NMR spectroscopic data of 1l matched those previously described.[28]
#
2-[(3-Pyridyl)amino]-1,2-diphenylethanone (6)
Reaction of thiourea 1l (34.66 g, 0.15 mol) and benzoin (31.81 g, 0.15 mol) in acetic acid (150 mL) for 10 h afforded 6. An analytically pure sample was obtained by recrystallization from EtOH/H2O (1:1).
Yield: 95% (40.9 g); mp 141–142 °C.
IR (ATR): 3326w, 2336w, 1678s, 1589s, 1578s, 1342s, 795s, 755s, 711s, 700s, 691s, 681s, 668s, 618s cm–1.
1H NMR (601 MHz, CDCl3): δ = 8.05 (d, J = 2.8 Hz, 1 H), 7.94–7.91 (m, 2 H), 7.86 (dd, J = 4.6, 1.3 Hz, 1 H), 7.49–7.45 (m, 1 H), 7.39–7.34 (m, 4 H), 7.22 (dd, J = 10.5, 4.8 Hz, 2 H), 7.16–7.12 (m, 1 H), 6.94 (dd, J = 8.3, 4.6 Hz, 1 H), 6.84 (ddd, J = 8.3, 2.9, 1.3 Hz, 1 H), 5.94 (d, J = 6.6 Hz, 1 H), 5.46 (d, J = 6.4 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 196.28, 142.02, 139.30, 136.92, 136.44, 134.71, 133.76, 129.25, 128.91, 128.77, 128.43, 128.15, 123.64, 119.65, 62.25.
Anal. Calcd for C19H16N2O: C, 79.14; H, 5.59; N, 9.72. Found: C, 79.51; H, 5.62; N, 9.81.
#
N,N′-Bis(2,4,6-trimethylphenyl)thiourea (1m)[29] [30]
According to GP A, reaction of 2,4,6-trimethylaniline (65.0 g, 0.5 mol) and carbon disulfide (30 mL, 0.5 mol), after recrystallization from EtOH (300 mL) afforded 1m.
Yield: 91% (70.9 g); mp 187–189 °C (Lit. 175 °C,[9b] 202–203 °C[29]).
IR (ATR): 3321m, 3250–3150br, 3100–2900br, 1519s, 1475s, 1246s, 1220s, 854m, 654m cm–1.
1H NMR (400 MHz, CDCl3): δ = 7.94 (s, 1 H), 6.99 (s, 2 H), 6.84 (s, 2 H), 6.47 (s, 1 H), 2.38 (s, 6 H), 2.30 (s, 3 H), 2.22 (s, 3 H), 2.18 (s, 6 H).
13C NMR (101 MHz, CDCl3): δ = 181.37, 139.17, 137.69, 137.29, 136.11, 133.16, 130.65, 129.79, 129.05, 21.01, 18.52, 18.10.
NMR spectroscopic data of 1m matched those previously described.[29] [30]
#
N,N′-Bis(2,6-diisopropylphenyl)thiourea (1n)[30] [31]
According to GP B, reaction of 2,6-diisopropylaniline (90%, 394 g, 2 mol) and carbon disulfide (91.4 g, 1.2 mol) in diglyme (1 L), afforded 1n.
Yield: 39% (153.8 g); mp 223–224 °C (Lit.[31] 242 °C).
IR (ATR): 3135m, 2959s, 1523s, 1463m, 1261s, 1234s, 800s, 481m cm–1.
1H NMR (601 MHz, CDCl3): δ = 8.80 (s, 1 H), 7.31 (t, J = 7.7 Hz, 1 H), 7.22–7.17 (m, 3 H), 7.05 (t, J = 6.0 Hz, 2 H), 6.32 (s, 1 H), 3.34 (hept, J = 6.8 Hz, 2 H), 2.96 (hept, J = 6.8 Hz, 2 H), 1.29 (d, J = 6.9 Hz, 6 H), 1.23 (d, J = 6.8 Hz, 6 H), 1.18 (d, J = 6.8 Hz, 6 H), 0.96 (d, J = 6.9 Hz, 6 H).
13C NMR (101 MHz, CDCl3): δ = 182.44, 148.14, 146.64, 132.74, 130.60, 130.11, 128.81, 124.23, 123.62, 28.89, 28.58, 26.01, 24.43, 23.84, 22.10.
NMR spectroscopic data for 1n matched those previously described.[30]
#
#
Acknowledgment
We are grateful to Prof. J. Christoffers from the Carl von Ossietzky University of Oldenburg for recording IR spectra and to Dr. O. Tok from the Institute of Inorganic Chemistry, Academy of Science of the Czech Republic for recording NMR spectra.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/s-0039-1690338.
- Supporting Information
-
References
- 1 Current address: C. Azap, Chemetall GmbH, Trakehner Str. 3, 60487 Frankfurt am Main, Germany.
- 2 Current address: A. Christoffers, Diapharm Analytics GmbH, Würzburger Str. 2, 26121 Oldenburg, Germany.
- 3a Herrmann W. A. Kocher C Angew. Chem., Int. Ed. Engl.; 1997, 36: 2163 ; Angew. Chem. 1997, 109, 2256
- 3b Bourissou D, Guerret O, Gabbai FP, Bertrand G. Chem. Rev. 2000; 100: 39
- 3c N-Heterocyclic Carbenes in Transition Metal Catalysis. Glorius F. Springer; Berlin: 2007
- 3d Diez-Gonzalez S, Marion N, Nolan SP. Chem. Rev. 2009; 109: 3612
- 3e Corberán R, Mas E, Peris-Marzá E. Eur. J. Inorg. Chem. 2009; 1700
- 3f Hopkinson MN, Richter C, Schedler M, Glorius F. Nature 2014; 510: 485
- 3g N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis. Nolan SP. Wiley-VCH; Weinheim: 2014
- 3h N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools. Diez-Gonzalez S. Royal Society of Chemistry; Cambridge: 2017
- 4 Occhipinti G, Bjørsvik H.-R, Jensen VR. J. Am. Chem. Soc. 2006; 128: 6952
- 5a Lübbe C, Dumrath A, Neumann H, Beller M, Kadyrov R. ChemCatChem 2014; 6: 105
- 5b Kadyrov R, Azap C, Weidlich S, Wolf D. Top. Catal. 2012; 55: 538
- 5c Kadyrov R, Rosiak A. Chemistry Today 2009; 27: 24
- 6a Jahnke MC, Hahn FE. In N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools . Diez-Gonzalez S. Royal Society of Chemistry; London: 2017: 1-41
- 6b Benhamou L, Chardon E, Lavigne G, Bellemin-Laponnaz S, Cesar V. Chem. Rev. 2011; 111: 2705
- 6c Zhong R, Lindhorst AC, Groche FJ, Kühn FE. Chem. Rev. 2017; 117: 1970
- 7a Herrmann WA, Goossen LJ, Artus GR. J, Köcher C. Organometallics 1997; 16: 2472
- 7b Arduengo AJ. III, Krafczyk R, Schmutzler R. Tetrahedron 1999; 55: 14523
- 7c Hirano K, Urban S, Wang C, Glorius F. Org. Lett. 2009; 11: 1019; and references cited therein
- 7d Lv T, Wang Z, You J, Lan J, Gao G. J. Org. Chem. 2013; 78: 5723
- 8a Schönherr H.-J, Wanzlick H.-W. Justus Liebigs Ann. Chem. 1970; 731: 176
- 8b Schönherr H.-J, Wanzlick H.-W. Chem. Ber. 1970; 103: 1037
- 8c Arduengo AJ. III, Goerlich JR, Krafczyk R, Marshall WJ. Angew. Chem. Int. Ed. 1998; 37: 1963; Angew. Chem. 1998, 110, 2062
- 8d Dowe AP, Li H, Pratt RC, Lohmeijer BG. G, Culkin DA, Waymouth RM, Hedrick JL. Chem. Commun. 2006; 2881
- 8e Ogle JW, Zhang J, Reibenspies JH, Abboud KA, Miller SA. Org. Lett. 2008; 10: 3677
- 8f Ogle JJ. W, Miller SA. Chem. Commun. 2009; 5728
- 8g Mehrotra KN, Singh G. Synthesis 1980; 1001
- 9a Schroeder DC. Chem. Rev. 1955; 55: 181
- 9b Azizi N, Khajeh-Amiri A, Ghafuri H, Bolourtchian M. Mol. Diversity 2011; 15: 157
- 9c Li Z, Liu D, Chen Y, Yin Y, Wang Z, Sun X. J. Chem. Res. 2016; 40: 515; and references cited therein
- 10 Sharma S. Synthesis 1978; 803
- 11a Dyson G, George HJ. J. Chem. Soc. 1924; 1702
- 11b Natarajan A, Guo Y, Arthanari H, Wagner G, Halperin JA, Chorev M. J. Org. Chem. 2005; 70: 6362
- 11c Štrukil V, Igrc MD, Fábián L, Eckert-Maksić M, Childs SL, Reid DG, Duer MJ, Halasz I, Mottilloe C, Friščić T. Green Chem. 2012; 14: 2462
- 12 Staab HA, Walther G. Justus Liebigs Ann. Chem. 1962; 657: 98
- 13a Ballabeni M, Ballini R, Bigi F, Maggi R, Parrini M, Predieri G, Sartori G. J. Org. Chem. 1999; 64: 1029
- 13b Venkatesh P, Pandeya SN. E-J. Chem. 2009; 6: 495
- 13c Maddani MR, Prabhu KR. J. Org. Chem. 2010; 75: 2327
- 14 Piel I, Pawelczyk MD, Hirano K, Fröhlich R, Glorius F. Eur. J. Org. Chem. 2011; 5475
- 15a Biltz H. Ber. Dtsch. Chem. Ges. 1907; 40: 4799
- 15b Klüpfel KW, Stumpf HR, Behmenburg H, Neugebauer W, Süß O, Tomanek M. German Patent Appl. DE 1060713, 1959 ; Chem. Abstr. 1961, 55, 20735b
- 16 Grimmett MR. In Science of Synthesis, Vol. 12. Neier R. Georg Thieme Verlag; Stuttgart: 2002
- 17 Pesch J, Harms K, Bach T. Eur. J. Org. Chem. 2004; 2025
- 18 Carpenter MS, Easter WM, Wood TF. J. Org. Chem. 1951; 16: 586
- 19 Dyson GM, George HJ, Hunter RF. J. Chem. Soc. 1927; 436
- 20 Pohloudek-Fabini R. Arch. Pharm. 1965; 298: 51
- 21 Strukil V, Gracin D, Magdysyuk OV, Dinnebier RE, Friscic T. Angew. Chem. Int. Ed. 2015; 54: 8440
- 22 Huebner CF, Marsh JL, Mizzoni RH, Mull RP, Schroeder DC, Troxell HA, Scholz CR. J. Am. Chem. Soc. 1953; 75: 2274
- 23 Pasha MA, Madhusudana Reddy MB. Synth. Commun. 2009; 39: 2928
- 24 Natarajan A, Guo Y, Arthanari H, Wagner G, Halperin JA, Chorev M. J. Org. Chem. 2005; 70: 6362
- 25 Lippert KM, Hof K, Gerbig D, Ley D, Hausmann H, Guenther S, Schreiner PR. Eur. J. Org. Chem. 2012; 5919
- 26 Kokorev GI, Yambushev FD. Zh. Obshch. Khim. 1987; 57: 1552
- 27 Deady LW, Ganame D, Hughes AB, Quazi NH, Zanatta SD. Aust. J. Chem. 2002; 55: 287
- 28 Katritzky AR, Witek RW, Rodriguez-Garcia V, Mohapatra PP, Rogers JW, Cusido J, Abdel-Fattah AA. A, Steel PJ. J. Org. Chem. 2005; 70: 7866
- 29 Yang D, Chen Y.-C, Zhu N.-Y. Org. Lett. 2004; 6: 1577
- 30 Findlater M, Hill NJ, Cowley AH. Dalton Trans. 2008; 4419
- 31 Walter W, Randau G. Justus Liebigs Ann. Chem. 1969; 722: 52
For reviews, see:
For selected references, see:
-
References
- 1 Current address: C. Azap, Chemetall GmbH, Trakehner Str. 3, 60487 Frankfurt am Main, Germany.
- 2 Current address: A. Christoffers, Diapharm Analytics GmbH, Würzburger Str. 2, 26121 Oldenburg, Germany.
- 3a Herrmann W. A. Kocher C Angew. Chem., Int. Ed. Engl.; 1997, 36: 2163 ; Angew. Chem. 1997, 109, 2256
- 3b Bourissou D, Guerret O, Gabbai FP, Bertrand G. Chem. Rev. 2000; 100: 39
- 3c N-Heterocyclic Carbenes in Transition Metal Catalysis. Glorius F. Springer; Berlin: 2007
- 3d Diez-Gonzalez S, Marion N, Nolan SP. Chem. Rev. 2009; 109: 3612
- 3e Corberán R, Mas E, Peris-Marzá E. Eur. J. Inorg. Chem. 2009; 1700
- 3f Hopkinson MN, Richter C, Schedler M, Glorius F. Nature 2014; 510: 485
- 3g N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis. Nolan SP. Wiley-VCH; Weinheim: 2014
- 3h N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools. Diez-Gonzalez S. Royal Society of Chemistry; Cambridge: 2017
- 4 Occhipinti G, Bjørsvik H.-R, Jensen VR. J. Am. Chem. Soc. 2006; 128: 6952
- 5a Lübbe C, Dumrath A, Neumann H, Beller M, Kadyrov R. ChemCatChem 2014; 6: 105
- 5b Kadyrov R, Azap C, Weidlich S, Wolf D. Top. Catal. 2012; 55: 538
- 5c Kadyrov R, Rosiak A. Chemistry Today 2009; 27: 24
- 6a Jahnke MC, Hahn FE. In N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools . Diez-Gonzalez S. Royal Society of Chemistry; London: 2017: 1-41
- 6b Benhamou L, Chardon E, Lavigne G, Bellemin-Laponnaz S, Cesar V. Chem. Rev. 2011; 111: 2705
- 6c Zhong R, Lindhorst AC, Groche FJ, Kühn FE. Chem. Rev. 2017; 117: 1970
- 7a Herrmann WA, Goossen LJ, Artus GR. J, Köcher C. Organometallics 1997; 16: 2472
- 7b Arduengo AJ. III, Krafczyk R, Schmutzler R. Tetrahedron 1999; 55: 14523
- 7c Hirano K, Urban S, Wang C, Glorius F. Org. Lett. 2009; 11: 1019; and references cited therein
- 7d Lv T, Wang Z, You J, Lan J, Gao G. J. Org. Chem. 2013; 78: 5723
- 8a Schönherr H.-J, Wanzlick H.-W. Justus Liebigs Ann. Chem. 1970; 731: 176
- 8b Schönherr H.-J, Wanzlick H.-W. Chem. Ber. 1970; 103: 1037
- 8c Arduengo AJ. III, Goerlich JR, Krafczyk R, Marshall WJ. Angew. Chem. Int. Ed. 1998; 37: 1963; Angew. Chem. 1998, 110, 2062
- 8d Dowe AP, Li H, Pratt RC, Lohmeijer BG. G, Culkin DA, Waymouth RM, Hedrick JL. Chem. Commun. 2006; 2881
- 8e Ogle JW, Zhang J, Reibenspies JH, Abboud KA, Miller SA. Org. Lett. 2008; 10: 3677
- 8f Ogle JJ. W, Miller SA. Chem. Commun. 2009; 5728
- 8g Mehrotra KN, Singh G. Synthesis 1980; 1001
- 9a Schroeder DC. Chem. Rev. 1955; 55: 181
- 9b Azizi N, Khajeh-Amiri A, Ghafuri H, Bolourtchian M. Mol. Diversity 2011; 15: 157
- 9c Li Z, Liu D, Chen Y, Yin Y, Wang Z, Sun X. J. Chem. Res. 2016; 40: 515; and references cited therein
- 10 Sharma S. Synthesis 1978; 803
- 11a Dyson G, George HJ. J. Chem. Soc. 1924; 1702
- 11b Natarajan A, Guo Y, Arthanari H, Wagner G, Halperin JA, Chorev M. J. Org. Chem. 2005; 70: 6362
- 11c Štrukil V, Igrc MD, Fábián L, Eckert-Maksić M, Childs SL, Reid DG, Duer MJ, Halasz I, Mottilloe C, Friščić T. Green Chem. 2012; 14: 2462
- 12 Staab HA, Walther G. Justus Liebigs Ann. Chem. 1962; 657: 98
- 13a Ballabeni M, Ballini R, Bigi F, Maggi R, Parrini M, Predieri G, Sartori G. J. Org. Chem. 1999; 64: 1029
- 13b Venkatesh P, Pandeya SN. E-J. Chem. 2009; 6: 495
- 13c Maddani MR, Prabhu KR. J. Org. Chem. 2010; 75: 2327
- 14 Piel I, Pawelczyk MD, Hirano K, Fröhlich R, Glorius F. Eur. J. Org. Chem. 2011; 5475
- 15a Biltz H. Ber. Dtsch. Chem. Ges. 1907; 40: 4799
- 15b Klüpfel KW, Stumpf HR, Behmenburg H, Neugebauer W, Süß O, Tomanek M. German Patent Appl. DE 1060713, 1959 ; Chem. Abstr. 1961, 55, 20735b
- 16 Grimmett MR. In Science of Synthesis, Vol. 12. Neier R. Georg Thieme Verlag; Stuttgart: 2002
- 17 Pesch J, Harms K, Bach T. Eur. J. Org. Chem. 2004; 2025
- 18 Carpenter MS, Easter WM, Wood TF. J. Org. Chem. 1951; 16: 586
- 19 Dyson GM, George HJ, Hunter RF. J. Chem. Soc. 1927; 436
- 20 Pohloudek-Fabini R. Arch. Pharm. 1965; 298: 51
- 21 Strukil V, Gracin D, Magdysyuk OV, Dinnebier RE, Friscic T. Angew. Chem. Int. Ed. 2015; 54: 8440
- 22 Huebner CF, Marsh JL, Mizzoni RH, Mull RP, Schroeder DC, Troxell HA, Scholz CR. J. Am. Chem. Soc. 1953; 75: 2274
- 23 Pasha MA, Madhusudana Reddy MB. Synth. Commun. 2009; 39: 2928
- 24 Natarajan A, Guo Y, Arthanari H, Wagner G, Halperin JA, Chorev M. J. Org. Chem. 2005; 70: 6362
- 25 Lippert KM, Hof K, Gerbig D, Ley D, Hausmann H, Guenther S, Schreiner PR. Eur. J. Org. Chem. 2012; 5919
- 26 Kokorev GI, Yambushev FD. Zh. Obshch. Khim. 1987; 57: 1552
- 27 Deady LW, Ganame D, Hughes AB, Quazi NH, Zanatta SD. Aust. J. Chem. 2002; 55: 287
- 28 Katritzky AR, Witek RW, Rodriguez-Garcia V, Mohapatra PP, Rogers JW, Cusido J, Abdel-Fattah AA. A, Steel PJ. J. Org. Chem. 2005; 70: 7866
- 29 Yang D, Chen Y.-C, Zhu N.-Y. Org. Lett. 2004; 6: 1577
- 30 Findlater M, Hill NJ, Cowley AH. Dalton Trans. 2008; 4419
- 31 Walter W, Randau G. Justus Liebigs Ann. Chem. 1969; 722: 52
For reviews, see:
For selected references, see: