
Subscribe to RSS
DOI: 10.1055/s-0039-1690762
Organo-f-Complexes for Efficient and Selective Hydroborations
Publication History
Received: 17 November 2019
Accepted: 18 November 2019
Publication Date:
02 January 2020 (online)

Abstract
Organo-f-complexes catalyzing small molecule transformations have been a hot topic in the past few years. Compared to other transformations, the hydroboration of C=X (X = C, N, O) unsaturated bonds serves as an important strategy to prepare organoborane derivatives, which are important intermediates in organic synthesis. This review outlines recent advances in organolanthanide and organoactinide complexes promoting the hydroboration of C=X containing substrates. After a brief introduction, three types of hydroboration will be presented: alkene hydroboration, carbonyl hydroboration, and imine and nitrile hydroborations. The catalytic performance, mechanism, and kinetic studies are discussed in detail, aiming to emphasize the catalytic differences between the diverse organo-f-catalysts. Additionally, challenges and future directions of this field are also presented.
1 Introduction
2 Alkene Hydroboration
3 Carbonyl Hydroboration
4 Imine and Nitrile Hydroboration
5 Conclusions and Outlook
#
Keywords
organolanthanide - organoactinide - hydroboration - selective - alkene - imine - ketone - aldehydeBiographical Sketches

Heng Liu obtained his B.Eng. degree in polymer material and engineering from Nanchang University in 2009, and Ph.D. in polymer chemistry and physics from Changchun Institute of Applied Chemistry, Chinese Academy of Science. In 2015, he was awarded a Guangdong-Technion Israel Institute of Technology (TGIIT) postdoctoral fellowship to work with Prof. Moris S. Eisen on novel organoactinide pre-catalysts. He is currently an Associate Professor at Changchun Institute of Applied Chemistry, Chinese Academy of Science. His current research interests include olefin/diene polymerization by early/late transition metal complexes and small molecule transformation by organo-f-element complexes.

Moris S. Eisen immigrated to Israel in 1977 from Colombia. He received his Ph.D. from the Hebrew University in Jerusalem under the supervision of Prof. Jochanan Blum. In 1990 as a Weizmann Postdoctoral Fellow, he joined Prof. Tobin J. Marks’ group at Northwestern University. In 1992 he was awarded the State of Israel Alon Fellowship and joined the Department of Chemistry at the Technion–Israel Institute of Technology (since 2007, Schulich Faculty of Chemistry). Since 2003, he is a Full Professor and has served from 2004 as the Head of the Institute of Catalysis Science and Technology at the Technion. He is incumbent of the Samuel O. Friedlander Academic Chair in Chemistry.
Introduction
Organo-f-element complex, including organolanthanides and organoactinides, catalyzed small molecule transformations have experienced colossal acceleration in research activity in the past few years.[1] The unique advantages of organo-f-element complexes, versus other main group, transition-metal complexes, lies in the presence of the f orbitals and large ionic radii, which leads to large coordination numbers, large and flexible coordination geometries, high Lewis acidity, and thereof distinctive catalytic behaviors.[2]
The catalytic performances of organo-f-element complexes in organic transformations are significantly influenced by the nature of metal ions and the steric and electronic effects of the ancillary ligands. For instance, for the majority of organolanthanide-promoted hydroelementations, larger metal ions bearing more sterically opening coordination spheres generally demonstrate higher activity than smaller counterparts,[3] therefore, the reactivities of organolanthanides can be easily fine-tuned according to the rule of the lanthanide contraction.[4] Moreover, lanthanides and early actinide (Th) exhibit predominantly one oxidation state (+3 and +4 respectively), excluding conventional oxidative addition/reductive elimination pathways, which is very common in transition-metal complexes.[5] Olefin-insertion and σ-bond metathesis are the two most general reactive patterns for organolanthanide- and organoactinide-promoted transformations.[4a]
Due to the highly oxophilic nature of the lanthanide and actinide centers, when reacting with oxygen-containing substrates, thermodynamically stable and catalytically inactive Ln–O and An–O bonds are preferably formed, making the transformation of these substrates extremely challenging.[5] [6] Therefore, despite the tremendous advances that we have witnessed in small molecule transformations, only a few of them have involved oxygen-containing substrates. Partially, for the same reason, organolanthanide- and organoactinide-catalyzed hydroboration have undergone a renaissance only in recent years due to the presence of oxygen atoms in some borane agents, such as catecholborane and pinacolborane.
Organoboranes are a class of vital organic intermediates in a variety of organic transformations, and hydroboration of unsaturated bonds serves as a powerful strategy in preparing organoboranes.[7] Ever since the milestone discovery of the hydroboration of the C=C bond by Brown and co-workers,[8] rapid development and continuous momentum has been gained in this area. Diversified main group metal catalysts (Li,[9] K,[10] Mg,[11] Al,[12] Zn[13]), transition-metal catalysts (Ti,[14] Co,[15] Ni,[16] Fe,[17] Pd,[18] Cu,[19] Rh,[20] Ir,[21] Ag,[22]), organocatalysts,[23] and even catalyst-free systems,[24] etc., were designed to efficiently and selectively promote hydroborations of unsaturated bonds. Reviews or chapters on the hydroboration of unsaturated bonds have been systematically compiled previously,[25] whereas few of them addressed organolanthanide and organoactinide systems, despite their indispensable significance in this field.
Hence, in this review, we will concentrate on recent advances in organolanthanide- and organoactinide-mediated hydroboration reactions. We mainly focus on three types of hydroborations, i.e. alkene hydroboration, carbonyl hydroboration, and imine and nitrile hydroboration, and we draw some conclusions on the challenges and future development directions of this field.
# 2
Alkene Hydroboration

Organo-f-complex mediated hydroboration reactions were firstly reported by the Marks group in 1992.[26] In this report, cyclopentadienyl lanthanum and samarium complexes 1–4 (Figure [1]) catalyzed the hydroboration using catecholborane as the borating agent, and the use of different types of alkenes was demonstrated. In the presence of a 25–100-fold excess of hex-1-ene, complex 1 exhibits an efficient catalytic rate (TOF ≈ 200 h–1), selectively affording the anti-Markovnikov product, hexan-1-ol, after the oxidative workup (Scheme [1]). Subsequent studies on various alkene substrates showed that the reaction rate followed the order of terminal alkene ≥ terminal disubstituted alkene > internal disubstituted alkene > internal trisubstituted alkene, and no reaction was detected for the tetrasubstituted substrate, 2,3-dimethylbut-2-ene. Similar to other organolanthanide-catalyzed small molecule transformations,[27] this phenomenon is likely caused by the steric demands at the metal center, i.e. sterically less congested substrates are more easily accessible to the active species. For the same reason, both larger metal ions and more sterically opening ligation were able to increase the hydroboration rate significantly. For instance, the reaction rate of complex 1 is 10-fold greater than that of complex 2, and complex 3 is fourfold as active as complex 2. The readily accessible samarium complex 4 also promoted the hydroboration process efficiently.

The catalytic mechanism proposed by the Marks group is presented in Scheme [2]. In the first activation step, the protonolysis of the lanthanide alkyl catalyst with catecholborane generates the active species, lanthanide hydride 2a, which is followed by the insertion of an olefin in an anti-Markovnikov manner to form complex 2b. Subsequent σ-bond metathesis with another molecule of catecholborane gives the final product, the boronate ester 2c, and simultaneously regenerates the lanthanide hydride.

Following this pioneering research, the Evans group extended the catalyst categories beyond lanthanide cyclopentadienyl complexes.[28] They successfully demonstrated that ancillary ligands were not strictly required for the hydroboration process. For the hydroboration reaction of dec-1-ene with catecholborane, samarium triiodide (SmI3), diiodosamarium tert-butoxide [(t-BuO)SmI2], and the homoleptic samarium isopropoxide [( i PrO)3Sm], were all found to be efficient catalysts, affording decan-1-ol in good yields after oxidation with hydrogen peroxide, whereas SmBr3, SmCl3, SmF3, and Sm(OTf)3 were inactive. Other trivalent group 3 and lanthanide salts, including ScI3, PrI3, LuI3, also displayed good activities towards the hydroboration process. Subsequent studies of the scope of the reaction revealed that mono-, di-, and trisubstituted alkene substrates could efficiently undergo hydroboration when being catalyzed by 10 mol% SmI3. For styrenic substrates, primary alcohol products, i.e. anti-Markovnikov products, resulted predominately; trisubstituted olefins, such as phenylcyclohexene and α-pinene, gave exclusively cis-addition products.
The possibility of the hydroboration of functionalized olefins was also explored. In the presence of 10 mol% SmI3, pent-3-en-1-ol was hydroborated with catecholborane in 73% yield, giving the two diol isomers in an 11:1 ratio (Scheme [3]).

Moreover, the regioselectivity of this SmI3-promoted hydroboration was discovered to be time dependent. For dec-1-ene hydroboration (Scheme [4], top), extending the reaction time from 3 hours to 18 hours resulted in a change of the ratio of the two products, decan-1-ol and decan-2-ol, from 3:1 to 50:1. A similar phenomenon was also reflected in the diastereoselectivity of two alcohol enantiomers (Scheme [4], bottom). These results can be explained by the isomerization towards the kinetically favored boronate esters with prolonged reaction times.

The Teuben group systematically explored the catalytic behaviors of a family of cyclopentadienyl and benzamidinate lanthanide complexes towards the hydroboration of hex-1-ene.[29] It was found that yttrocene complexes displayed much lower catalytic activities than their lanthanocene counterparts because of the smaller atom radius and thereof increased σ-bond metathesis transition state energy (formation of the hydroboration product and regeneration of metal hydride is the rate-determining step). For instance, complex 5 was found to be less active than complex 1 under identical conditions. This conclusion is in line with Marks’s previous hydroboration observations and other lanthanide-catalyst-promoted catalytic transformations.[26] Changing the metal attached alkyl substituents from bis(trimethylsilyl)methyl (5) to methyl (6) and hydride (7) gave similar results to complex 5. Employing sterically less congested yttrocene complexes, 8 and 9, revealed remarkable increases in catalytic activities.
Besides cyclopentadienyl complexes, benzamidinate yttrium counterparts were also investigated by the Teuben group, based on the consideration that this type of ligand is a ‘harder’ Lewis base, which will have a strong influence on the transition state of the final σ-bond metathesis rate-determining step. Furthermore, the steric bulkiness of benzamidinate ligands is presumed to be between that of the cyclopentadienyl and pentamethylcyclopentadienyl ligands, which favors building the relationship between steric hindrance and catalytic performances. Through the catalytic results, it was revealed that all benzamidinate complexes are active in the hydroboration process, but their activities are roughly 1/30 to 1/12 of that of the lanthanocene complex 1. Complexes 10 and 11 (Figure [2]) showed catalytic activities substantially higher than 5–7, but lower than 8 and 9, which is in accordance with the steric characteristics. When Lewis bases are present in the complex, such as THF in 12, rapid decomposition of catecholborane to bis(pinacolato)diboron dimer is detected.

Livinghouse and co-workers, carried out alkene-pinacolborane hydroborations using commercially available tris[bis(trimethylsilyl)amide]lanthanum (13) and cyclopentadienyl yttrium 14 complexes.[30] In the presence of 3 mol% of 13, hex-1-ene was hydroborated by pinacolborane (HBpin) giving hexan-1-ol in 90% isolated yield, which is significantly superior to that obtained using complex 14 under identical conditions. The hydroboration was also highly efficient for other alkene substrates, including cyclohexene, indene, styrenic monomers, all of which proceeded to >99% conversions and revealed anti-Markovnikov selectivity. It is of note that the introduction of strong electron-withdrawing or -donating para substituents to styrenic substrates had little influence on the catalytic behaviors, both types of compounds were catalyzed rapidly under mild conditions.
Due to steric influences, the reactivity of alkene substrates in hydroboration transformations usually follows the order of mono- > di- > tri- > tetrasubstituted C=C bonds. However, Villiers and Ephritikhine found that in the presence of UCl4 or NdCl3 the hydroboration of alkenes with LiBH4 followed the completely opposite order to that in previous reports.[31] Treating 2,3-dimethylbut-2-ene with LiBH4 and UCl4 gave uranium alkylborohydride in high yield (90%), which immediately gave 2,3-dimethylbutan-2-ol upon oxidation by alkaline hydrogen peroxide (Scheme [5]). Trisubstituted alkenes, such as α-pinene, 2-methylpent-2-ene, etc., were less reactive than 2,3-dimethylbut-2-ene and the reactions required a longer time to reach completion. Most striking is that less substituted alkenes (cyclohexene, 2-methylpropene, and hex-1-ene) were found to be inert towards the hydroboration process, which completely disobeys the general rule concluded previously. NdCl3 was also found to demonstrate similar results, but with slower reactivities.

Inspired by lanthanocene-mediated cyclization/silylation of 1,1-disubstituted dienes for the preparation of functionalization carbocycles, Molander and Pfeiffer reported the first examples of an organolanthanide-catalyzed cyclization/boration reaction of 1,5- and 1,6-dienes to give primary cyclic alcohols.[32] Initially they examined the reaction between hexa-1,5-diene and catecholborane catalyzed by complex 4, which proved ineffective. Changing to other lanthanide metallocene complexes 14 and 15 and using HBpin as the hydroboration reagent gave the desired cyclized cyclopentylmethanol in yields up to 50%, but with many byproducts, such as hex-5-en-1-ol and hexane-1,6-diol, which came from σ-bond metathesis in an undesired uncyclized hydroboration fashion (Scheme [6]). In order to slow down σ-bond metathesis and simultaneously promote the cyclization reaction, the reaction of 1,3-dimethyl-1,3-diaza-2-boracyclopentane (1,3-dimethyl-1,3,2-diazaborolidine) with hexa-1,5-diene was examined in the presence of complex 4, and it then proved to be an efficient and selective hydroboration reagent affording the cyclic alcohol product in 86% yield (Scheme [7], R = H, n = 1). Whereas, the yttrocene complex 14 did not show any activity in this reaction perhaps due to the smaller metal ionic radius as concluded above. Increasing the steric effects on the ancillary cyclopentadienyl ligands by using complexes 15–17 (Figure [2]), the desired products were obtained in yields ranging from 40% for Ln = Lu (16) to 62% for Ln = Y (15), and 74% for Ln = Sm (17). The use of other substituted 1,5- and 1,6-dienes was also feasible in this cyclization/boration reaction, but the formation of six-membered rings is comparatively less efficient than the formation of five-membered rings (Scheme [7]).

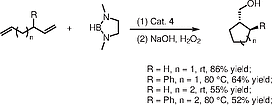
The proposed mechanism for the cyclization/boration reaction is presented in Scheme [8]. The precatalyst firstly reacts with the organoborane to generate the catalytically active species lanthanide hydride 8a, which then regioselectively inserts into one of the terminal double bonds to give metal hydrocarbyl compound 8b. Subsequently, intramolecular cyclization between the metal species and another double bond followed by a σ-bond metathesis with another molecule of the organoborane gives the desired cyclic product 8d and simultaneously regenerates the lanthanide hydride species.

Koga and Kulkarni investigated Cp2SmH-catalyzed ethylene hydroboration by ab initio MO methods.[33] In their calculations, ethylene coordinates to the samarium center to form a π-complex in the first step. Subsequent insertion of the ethylene molecule into the Sm–H bond through an energy barrier of 4.2 kcal/mol gives intermediate Cp2SmC2H5, which could coordinate with a borane reagent HB(OH)2 to yield a samarium metallacycle intermediate bridged with hydride and OH groups, respectively. Releasing the product C2H5B(OH)2 is the final step, which also gives back the original active species, this step is also calculated as the rate-determining step.
# 3
Carbonyl Hydroboration

Hydroboration of carbonyl compounds serves as an efficient strategy to access primary and secondary alcohols (Scheme [9]). Comparing to reductions by active hydride reagents, such as NaBH4, LiBH4, etc., carbonyl hydroboration generally displays good functionality tolerance, highly catalytic efficiency, outstanding selectivity, etc., and thus it has attracted great attention in the past few years.[11k] [25a] [34] The first examples of organo-f-element complex mediated carbonyl hydroboration was reported by the Marks group in 2017, in which the rapid, clean hydroboration of ketones and aldehydes with HBpin was achieved in the presence of the homoleptic lanthanide amide complexes 13 and 18–20 (Figure [3]).[35] The reaction of benzophenone catalyzed by 0.01 mol% of complex 13 gave ~99% of the hydroboration product in less than 5 minutes; the TOF was measured to be 40,000 h–1.

Complexes 18–20 also revealed high efficiencies, but with relative smaller reactivities. Subsequent scope studies by using complex 13 demonstrated that the hydroboration of both electron-rich aliphatic ketones and less electron-rich aromatic ketones proceeded smoothly, but that the former required lower catalyst loadings (0.01 mol% vs 0.1 mol%). High selectivity and good functionality compatibility were also demonstrated from the ketone hydroboration reaction with various types of functional groups, such as halogens, nitro, alkenes, alkynes, etc., which successfully survived during the reaction, and HBpin exclusively inserts into the carbonyl bond. Besides ketones, aldehydes can also be cleanly reduced.[25a]
The rate laws for ketone and aldehyde hydroboration were respectively determined by using dicyclohexyl ketone and cyclohexanecarbaldehyde as substrates. For ketone hydroboration, first-order dependence on catalyst, ketone, and HBpin was revealed (Equation 1), with activation parameters of ΔH ‡ = 17 ± 1 kcal/mol and ΔS ‡ = –15 ± 2 e.u., respectively. For the aldehyde hydroboration, first-order dependence on precatalyst and zero-order dependence on aldehyde and HBpin was observed (Equation 2), with thermodynamic activation parameters of ΔH ‡ = 12 ± 2 kcal/mol and ΔS ‡ = –33 ± 7 e.u., respectively. The large enthalpy ΔH ‡ in ketone hydroboration reflected unfavorable steric repulsions in the transition state. Therefore, when an equimolar mixture of acetophenone and benzaldehyde was reacted, a preference for aldehyde hydroboration was observed, with 91% conversion for benzaldehyde and 9% conversion for acetophenone. In a similar competition experiment, the reaction of 4-acetylbenzaldehyde with 1.0 equivalent HBpin was studied, and preferential hydroboration of the aldehyde (91%) was again observed (Scheme [10]).



Xue, Shen, and co-workers also employed complex 13 to promote ketone and aldehyde hydroborations, and substantially high catalytic efficiencies were revealed.[36]
In 2017–2018, Xue, Bao, Shen, and co-workers reported that homoleptic cyclopentadienyl lanthanide complexes 24–29 (Figure [3]) served as excellent catalysts for the hydroboration of carbonyl compounds with HBpin.[37] The reduction process can be carried out in various organic solvents, including toluene, CHCl3, and CH2Cl2, and even THF, DME, and 1,4-dioxane. This has great advantages compared to other organo-f-element complex mediated transformations as THF, DME, and 1,4-dioxane are generally considered as solvent-poisons to organo-f-element catalysts. Similar to Marks’ report, complexes 24–29 also displayed good functionality tolerances, and they can selectively hydroborate the carbonyl bond while not reacting with other functional groups, such as halides, nitro group, etc. High chemoselectivity was also revealed from these systems, even at high conversions of the carbonyl group, where other unsaturated bonds, such as cyano, alkyne, alkene, remained intact throughout the reaction. Perhaps due to the less electrophilic and sterically more congested carbonyl center in ketones, they generally displayed sluggish reactivities compared to their aldehyde counterparts, this is consistent with other transition metal and main group metal complexes,[12g] [34c] but opposite to the results obtained for lanthanide amide complexes.[35] Also for steric reasons, in competitive aldehyde/ketone hydroboration studies (similar to Scheme [10]) mediated by complex 28, aldehydes are preferentially reduced rather than ketones under identical conditions. During kinetic studies, it was found that the rates follow first-order dependence on aldehyde/ketone, HBpin, and complex 28, respectively, giving the kinetic rate law shown in Equation 3.

Xue, Shen, and co-workers also utilized the β-diimide bivalent rare-earth borohydride complexes 30 and bis(β-diimide) rare earth amide complexes 31 to catalyze the hydroboration between borane reagents and carbonyl compounds. The reactions rapidly, mildly, and efficiently gave the corresponding alcohol products.[38]
In 2018, Ma and co-workers, reported the hydroboration of aldehydes and ketones by the well-defined Schiff base heavy rare-earth ytterbium iodide complex 32.[39] Similar to other light rare-earth complexes, this complex also displayed good functionality tolerances, and the presence of electron-withdrawing and -donating groups showed little influence on the hydroboration rate; the target pinacolborate esters were obtained with high conversions in very short reaction times. Hydroboration of more sterically bulky ketone substrates, as expected, required higher catalyst loadings than those of aldehyde substrates, reflecting a sluggish reactivity therein. Under identical conditions, it was found that complex 32 exhibited higher activity in the hydroboration of aldehydes, such as α,β-unsaturated cinnamaldehyde, than the lanthanide amide complex 13 and cyclopentadienyl lanthanide complex 28.
Lanthanide aryloxides 33–38 (Figure [4]) were evaluated as catalysts by Xue, Bao, and co-workers in the hydroboration of carbonyl compounds with HBpin.[40] It was found that the complexes containing bulkier ligands have superior activities compared to their sterically less hindered counterparts. For instance, reducing the steric hindrance of ortho-substituents from tert-butyl (34) to isopropyl (35) and methyl (36) resulted in a reduction in alcohol conversions from 77% to 58%. Moreover, for lanthanides bearing the same ligands, the central Ln metals demonstrated that the order of catalytic reactivity follows Nd (38) > Sm (37) > Y(34) ~ Yb (33), with complex 38 as the optimal catalyst. In the presence of 0.05 mol% of complex 38, most aromatic aldehydes were fully converted in 10–30 min, and functional groups, including heterocycles, halides, hydroxy, etc., showed a small influence on the reaction; the corresponding alcohols can be achieved with high catalytic efficiencies. Beside aldehydes, ketones can also be hydroborated smoothly by complex 38, but with relatively lower reactivity, which is consistent with other lanthanide-mediated systems.

This superior reactivity of aldehydes over ketones was computationally evaluated, in which the energy barrier for the rate-determining step for ketones is ca. 5 kcal/mol higher than that of the aldehydes, thus preferable hydroboration of aldehydes was generally observed.
Stereoselective hydroboration/reduction of ketones employing efficient catalysts bearing chiral ligands has been rarely investigated.[41`] [b] [c] [d] In 2018, Yao, Zhao, and co-workers reported the only example of an enantioselective reduction of ketones catalyzed by rare-earth metal complexes with phenoxy modified chiral prolinols (Scheme [11], Figure [4]).[41e] This reaction was catalyzed by the in situ formed complexes from 20% mol of the phenoxy-functionalized chiral prolinol ligand L1 and 10 mol% of the lanthanide amide precursors 13 and 21–23. The hydroboration reaction proceeded smoothly to afford the reduced alcohols in high yields. Additionally, it was found that lanthanide complexes with a central metal of moderate ionic radii, such as ytterbium amides 21, resulted in much higher ee values (40%), than lanthanide counterparts with larger (28% ee for 13 and 31% ee for 22) or smaller ionic radii (8% ee for 23). This is perhaps due to the size of the ligand matching well with the appropriate ionic radii of the ytterbium metal, which plays a pivotal role in controlling the enantioselectivity of the active species. Varying ligand frameworks L1–L6 (Figure [4]), together with precursor 21 (Figure 3), demonstrated that the phenoxy ligand is essential for the enantioselectivity, and bulkier substituents, such as tert-butyl at the ortho-position, generally resulted in higher ee values. Well-defined ytterbium complex 39 was also evaluated as a catalyst, due to its structural similarity to the in situ formed active species, and better conversions (up to 96%) and ee value (up to 81%) were observed.


Substrate scope studies on various substituted acetophenones showed that enantioselective reductions can be performed smoothly with complex 39. The presence of both electron-donating and -withdrawing groups gave the targeted alcohols in high yields (Scheme [12]), but with varying ee values. In general, it was concluded that substrates bearing ortho-substituents gave good-to-excellent enantioselectivities, while meta- or para-substituted counterparts resulted in relative lower ee values. The enantioselective reduction of an α,β-unsaturated ketone, chalcone, was also examined. In the presence of complex 40, the corresponding alcohol was obtained with up to 99% conversion and 83% ee; the double bond remained intact throughout the hydroboration (Scheme [13]).

In 2019, Marks, Lohr, and co-workers reported an efficient, highly active, and selective homogeneous catalyst 13 for ester reduction with HBpin.[42] In the presence of 1 mol% of 13, various types of esters were reduced in near quantitative yields at 25 °C or 60 °C, affording the target hydroborated alcohol products (Scheme [14]). Steric impediments at the alkoxy position (R′) significantly depress the reactivity, for instance, substrates with tert-butyl acetate required 16 hours to complete the reaction, whereas cyclohexyl or ethyl acetate required only 1 hour and 10 minutes, respectively. Comparatively, steric impediments at the acetyl position (R) displayed little influence on the reactivity. Besides steric effects, a significant increase in turnover was observed for substrates with electron-withdrawing substituents at the R position. Additionally, the charge density on the alkoxy group also played a pivotal role in determining the reactivity; the presence of a phenyl group at the R′ position reduced the rate dramatically. During the reduction, side reactions with nitro and alkenyl groups were not detected, demonstrating a high selectivity for the reaction. Competitive reactions between ester and oct-1-ene or oct-1-yne resulted exclusively in the reduction of the ester group.

Kinetic investigations for the ester hydroboration revealed first-order dependence on catalyst 13, and zero-order dependence on both HBpin and the ester substrate, giving the rate law shown in Equation 4. Based on stoichiometric reactions and DFT calculations, a probable mechanism is proposed in Scheme [15]. Activation of the ester coordinated complex 15a with HBpin, followed by coordination of another molecule of HBpin, gives the hemiacetal active species 15b. The HBpin molecule in this intermediate promotes La–O bond dissociation and B–O bond formation to generate intermediate 15c, which is simultaneously stabilized by one molecule of ester. Intramolecular rearrangement yields product R′OBpin, followed by coordination of another molecule of HBpin to give intermediate 15d. Final hydride transfer from the boron atom of the hydroborate–La complex to the coordinated ester 15e regenerates the active species 15a, and concurrently release the product of RCH2Bpin.


Different from other metal complexes, transforming oxygen-containing substrates is a great challenge for organoactinides due to the high oxophilicity of the actinide center.[1`] [e] [f] , [5] By using strongly basic and highly nucleophilic imidazolin-2-iminato ligands, the Eisen group found that organoactinides can successfully transform oxygenated substrates, such as by the Tishchenko reaction,[43] ring-opening polymerization of cyclic ester,[44] insertions of alcohol into carbodiimides,[6d] [e] [45] etc. In 2018, the Eisen group reported an unprecedented chemoselective hydroboration of ketones and aldehydes catalyzed by the actinide complexes 41 and 42 (Figure [4]).[46] This study not only demonstrated again the feasibility of transforming oxygen-containing substrates, but also investigated the possibility of regenerating an An–H bond from a thermodynamically stable An–O bond. In the presence of 0.1 mol% to 0.004 mol% of the complexes 41 and 42, carbonyl compounds were hydroborated with HBpin in almost quantitative conversions in 15 minutes, and with TOFs as high as 100,000 h–1, which is highest value ever reported for a hydroboration process. Despite of the high oxophilicity of the metal center, complex 41 revealed good tolerance towards different types of functional groups, such as amine, imine, halides, nitro group, ester, etc., and afforded the corresponding alcoholic boronate esters in high yields. Furthermore, no hydroboration occurred on other unsaturated bonds, such as alkenes, alkynes, or the cyano group, even in the presence of excess HBpin, demonstrating the high chemoselectivity for this system. It is important to note that the present catalytic system displays a ‘living’ behavior, and in three consecutive runs of adding substrates into the same reaction mixture, high catalytic reactivities were always successfully retained.
Based on stoichiometric reactions, a plausible mechanism was proposed (Scheme [16]) in which the actinide monohydride intermediate 16a, generated from the protonolysis of complex 41, serves as the active species. This species further inserts into the carbonyl moiety to form the corresponding alkoxo complex 16b. Subsequent σ-bond metathesis, with HBpin, gives the boronate ester product and regenerates simultaneously the active species 16a. The kinetic rate law measured indicates a first-order dependence on catalysts, HBpin, and the carbonyl motif. Activation parameters were determined from the Eyring and Arrhenius plots, with values of 25.4 (0.8) e.u. for ΔS ‡, 13.7(0.7) kcal/mol for ΔH ‡, and 14.3(0.7) kcal/mol for E a. Deuterium isotope studies using DBpin revealed that the reaction exhibited a KIE (k H/k D) of 2.51 (0.07), indicating that the hydride transfer is the rate-determining step.

# 4
Imine and Nitrile Hydroboration
Reduction of imines and nitriles is a crucial methodology to access primary and secondary amines, which are extensively present in natural products, drugs, polymers, and other industrial and academic encounters. Compared to direct reduction by hydrogenation,[47] or alkaline metal hydrides,[48] the hydroboration generally displays a high efficiency, and a high selectivity under mild conditions, and thus it has attracted much academic interest in over the past few years. To date, diverse Co,[49] Mg,[11f] [j] Ru,[50] Mo,[51] Au,[52] etc., complexes have been extensively studied for the hydroboration transformation; organo-f-element catalysts, however, are relatively less utilized. The first report on the organolanthanide-mediated hydroboration of a C=N moiety appeared in 2014 by the Marks group, in which pyridines were regioselective 1,2-dearomatized by lanthanum hydride complex 43 to give 1,2-dihydropyridines (Figure [5]).[53] Compared to previous dearomatization catalytic systems,[11l] [54] the advantages of this research include the employment of earth-abundant, low toxic, low-cost lanthanide complexes that have high atom-efficiency and high 1,2-regioselectivity at low catalyst loading (1 mol%) under mild conditions (Scheme [17]). Furthermore, good functional group compatibility was demonstrated for this system, despite the highly electrophilic nature of the lanthanide center. A wide range of pyridines possessing different types of functionalities, such as halides, methoxy, aryl groups, etc., underwent efficient hydroborations to give the target dearomatized products in moderate to high yields. Both electronic and steric factors were found to significantly influence the catalytic activities. The presence of electron-withdrawing groups led to increased hydroboration rates, and conversely, the presence of electron-donating groups resulted in lower TOFs. Besides pyridines, other benzofused N-heterocycles, including quinoline, isoquinoline, were hydroborated rapidly, affording the corresponding 1,2-dearomatized products in high yields.


Kinetic studies revealed that the reaction rate was first order for the concentration of catalyst 43, and inverse first order for HBpin. For pyridine concentration, however, the reaction rate law was first order when it was below 0.2 M, while zero-order at higher concentrations (Equation 5). The thermodynamic activation parameters, determined from the Eyring and Arrhenius plots, were ΔH ‡ = 15.7(0.5) kcal/mol, ΔS ‡ = –27.2(0.3) cal/mol, and E a = 14.3(0.7) kcal/mol.

A plausible mechanism for this hydroboration process is proposed (Scheme [18]) based on the stoichiometric reaction and DFT calculations. In the presence of pyridine, the dimeric complex 43 is firstly cleaved into the monomeric adduct Cp*2LaH(Py) (18a), which then could be coordinated by a second molecule of pyridine to form the bispyridine adduct Cp*2LaH(Py)2 (18b). This bispyridine adduct undergoes La–H 1,2-insertion across the aromatic C=N moiety to afford a Cp*2La(NC5H6)(Py) complex 18c, which is further identified as one of the two rate-determining steps by DFT calculations. In the presence of a molecule of coordinated HBpin, La–N/H–B σ-bond metathesis quickly occurs, and releases the final desired N-borylated 1,2-dihydropyridine, with simultaneous regeneration of 18a. During the catalytic process, a rapid equilibrium between 18a and 18f was also established, which represents a deactivation pathway during the catalytic process.

In 2018, Wang and co-workers reported the hydroboration of a large number of imines and nitriles using amide-functionalized N-heterocyclic carbene rare earth complexes 44–47 as catalysts.[55] They found that these complexes displayed high catalytic activity for the hydroboration of N-benzylideneaniline affording the desired hydroborated product in high yields. Under identical conditions, complexes 44–47 also demonstrated much better reactivities than other amido metal complexes, such as NaN(SiMe3)2, KN(SiMe3)2, Gd[N(SiMe3)2]3, [(Me3Si)2N]3RE(μ-Cl)Li(THF)3 (RE = Er, Y, Dy), indicating the important role of the carbene moiety in enhancing the catalytic activity. Studies of the substrate scope by varying R1 and R2 (Scheme [19], top) revealed that substituents with different electronic properties, such as alkyl, halides, methoxy, naphthyl, and hydroxy, showed little influence on the yields of the desired products. The reaction performed with the strong electron-withdrawing nitro group was an exception, in which a relatively poor yield resulted due to the coordination of the nitrogen to the rare earth metal. Subsequent substrate expansion to nitrile compounds revealed that, in the presence of 3 equivalents of HBpin and 2 mol% of catalyst, aromatic and aliphatic nitriles can be double hydroborated, regardless of the electronic and steric effects, to afford the dihydroborated products in excellent yields (89–99%) (Scheme [19], bottom).

A series of competition reactions among different C=O, C=N, C≡N, CO2Et, and C=C groups were also carried out. The reactivity was found to follow the order of C=O >> C=N > C≡N > CO2Et > C=C. Hence, when a mixture of ethyl 4-methylbenzoate and N-benzylideneaniline was hydroborated, the exclusive hydroboration of the imine was observed [Scheme [20], (1)]. For a substrate bearing both the cyano and imine groups, selective hydroboration on the imine was observed when 1.2 equivalents of HBpin were utilized, however, increasing the amount of HBpin to 4.5 equivalents led to a complete reduction of both imine and cyano groups [Scheme [20], (2)].

A plausible mechanism for the hydroboration of imines is presented as Scheme [21]. In the presence of HBpin, the rare-earth metal hydride species 21a is firstly generated, which subsequently inserts into a coordinated C=N imine bond to give the rare-earth amido intermediate 21c. This intermediate reacts with another molecule of HBpin to release the desired hydroborated product 21d and regenerate the active species 21a.

Xue, Bao, and co-workers also reported that complex 28 (Figure 3) was an effective catalyst for the hydroboration of imines in addition to carbonyl substrates.[37b] In the presence of 1 equivalent of HBpin and 2 mol% of complex 28 at 60 °C, aromatic imine derivatives containing an electron-withdrawing group (4-F) or an electron-donating group (4-MeO) delivered medium to good yields of the target products (Scheme [22]).

Motivated by recent reports of organolanthanide-catalyzed hydroboration of C=N and C≡N moieties, the Eisen group developed a series of organoactinide complexes to perform similar transformations. Benzonitrile underwent double hydroboration catalyzed by the thorium metallacycle complex 49 (0.1–1 mol%) in a short reaction time to afford the dihydroborated amine in high yields.[56] This reaction was later expanded to other nitrile derivatives, and it was found that electron-rich aromatic nitriles bearing electron-donating groups such as 4-Me, 4-MeO, etc., displayed higher reactivities than electron-withdrawing counterparts for which longer reaction times or larger catalyst loadings were required in order to completely transform the nitrile reagents. Polyaromatic nitrile compounds, such as 1-naphthonitrile, showed lower reactivity compared to benzonitrile. However, heteroatom-containing aromatic nitriles, such as furanacetonitrile, thiopheneacetonitrile, pyridine-4-acetonitrile, underwent the double hydroboration efficiently affording the corresponding compounds almost quantitatively. Moreover, different from known catalysts, for less reactive for aliphatic nitriles, complex 49 showed remarkable turnover frequencies for aliphatic nitrile compounds, which are comparable to aromatic counterparts, demonstrating the unique advantage of the organoactinide complexes.
Kinetic measurements for the double hydroboration of benzonitrile revealed first-order dependence on the catalyst 49, second-order dependence on HBpin, and zero-order dependence on benzonitrile, giving an equation as shown in Equation 6. Activation parameters were determined from the Eyring and Arrhenius plots with ΔS ‡, ΔH ‡, and E a values of –47.25(1.23) e.u., 6.99(0.44) kcal/mol, and 7.66(0.44) kcal/mol, respectively.

A plausible mechanism for the double nitrile hydroboration was proposed (Scheme [23]). The activation of the catalytic cycle is firstly achieved by the protonolysis of complex 49 to generate the thorium hydride species 23a, which then quickly inserts into the C≡N bond, allowing the formation of intermediate 23b. In the presence of HBpin, 23b is able to undergo σ-bond metathesis to release the monohydroborated imine product 23c, which quickly participates in the second catalytic cycle by coordinating back to active species 23a. The Th–H moiety in 23a quickly inserts into the C=N bond of 23c, giving the intermediate 23d bearing the dihydroborated amido substituent. In the presence of another molecule of HBpin, 23d undergoes Th–N/HBpin σ-bond metathesis to give the final double hydroborated amine product 23e, and concurrently regenerates the active species 23a. Deuterium isotope analysis suggested that the final hydroboration step is the turnover-limiting step for the catalytic process.

In addition to nitriles, complex 49 was also utilized in the hydroboration of imines.[56] Whereas the thorium complex 50 (Figure [5]), coordinated by the seven-membered N-heterocyclic iminato ligand, showed much better catalytic efficiency. Moreover, complex 50 displayed large substrate scope capabilities, a large number of aldimines, including ketimines, were hydroborated to afford the corresponding products in almost quantitative yields. Aldimines comprising electron-donating functionalities increased the reactivities substantially, in comparison to the electron-withdrawing counterparts. Kinetic studies on the PhC=NPh/HBpin/50 system revealed that the kinetic rate law follows has a first-order dependence on catalyst 50 and HBpin, and zero-order dependence on imine, giving rise to Equation 7.

The selective monohydroboration of carbodiimides is of great interest for scientists because the produced amidinates play a pivotal role in coordination chemistry. In 2018, the Eisen group reported a highly efficient and highly monoselective organoactinide-catalyzed hydroboration of carbodiimides using complexes 48 and 49 and 51–56 (Figure [5]).[57] Different from the previously reported magnesium systems,[11e] [g] the organoactinide complexes required lower catalyst loadings and shorter reaction times and exhibited, unprecedentedly, a monoselective reactivity (Scheme [24]). Compared to the actinide amido complexes 48 and 49 and 51–54, the actinide methyl counterparts displayed superior reactivities, affording the corresponding hydroborated products with higher yields under identical reaction conditions. Subsequently, the substrate scope revealed that steric properties played a significant role in determining the catalytic efficiency. For symmetrical carbodiimides bearing smaller groups, such as isopropyl, cyclohexyl, phenyl, etc., quantitative conversion can be achieved in shorter reaction times, while sterically congested counterparts require much longer reaction times to obtain a high conversion. Electronic properties showed a very small influence on the catalytic activities, affording the product in high yields.

An interesting phenomenon was observed when using unsymmetrical carbodiimide substrates. For the hydroboration of N-mesityl-N′-phenylcarbodiimide (MesNCNPh) and N-(2,6-diisopropylphenyl)-N′-phenylcarbodiimide (DippNCNPh), the Bpin group is selectively attached to the steric bulky side of the carbodiimide, rather than the steric opening side, affording N-{Bpin}-N-diisopropylphenyl-N′-phenylformamidine and N-{Bpin}-N-mesityl-N′-phenylformamidine as the sole reaction products (Scheme [25]).

The thermodynamic activation parameters, determined from Eyring and Arrhenius plots, were E a = 16.6(9) kcal/mol, ΔH ≠ = 16.0(3) kcal/mol, and ΔS ≠ = –27.7(6) e.u., respectively. Kinetic studies on the i PrNCN i Pr/HBpin/56 system revealed a first-order dependence on HBpin, i PrNCN i Pr, and 56, respectively, giving rise to the kinetic rate law as presented in Equation 8.

A plausible mechanism is proposed, based on stoichiometric reactions (Scheme [26]). Firstly, a rapid σ-bond metathesis between the actinide methyl complex and HBpin occurs to yield the catalytic active species 26a, which then inserts into the C=N bond of the carbodiimide substrate allowing the formation of actinide amidinate intermediate 26b. In the presence of another molecule of HBpin, the monohydroborated carbodiimide 26c is released as the final product with the concomitant regeneration of the active species 26a.

Also in 2018, the Eisen group reported another example of a catalytic dearomatization/hydroboration of N-heteroaromatics utilizing the thorium complexes 57 and 58.[58] In this research, complexes 57 and 58 displayed high catalytic activity in the hydroboration of pyridines, affording the 1-boryl-1,2-dihydropyridine product in high yields. Moreover, no 1,4-hydroborated product was detected during the reaction process, demonstrating a high 1,2-selective fashion (Scheme [27]). Subsequent substrate studies revealed a large scope capability, and pyridines with different types of meta- and para-substituted groups, including alkyl, aryl, halides, nitro, methoxy, etc., were hydroborated by HBpin selectively affording the corresponding products in moderate to high yields.

Furthermore, other N-heteroaromatic compounds, such as quinoline, pyrazine, pyrimidine, triazine, etc., were also feasible for the hydroboration process, affording the mono-, di-, and trihydroborated products in high yields. It is important to note that the remaining double bond in the hydroborated pyridine product remained intact during the reaction, indicating a highly chemoselective fashion of the precatalysts.
Kinetic investigations of the pyridine/HBpin/58 catalytic system revealed a first-order dependence on the concentration of pyridine, HBpin, and complex 58, respectively, giving rise to the kinetic rate as shown in Equation 9. The thermodynamic activation parameters, determined from the Eyring and Arrhenius plots, are E a = 20.3(1) kcal/mol, ΔH ‡ = 19.6(5) kcal/mol, and ΔS ‡ = –23.5(1) e.u, respectively.

A plausible catalytic mechanism was proposed (Scheme [28]). The activation of the cycle is firstly achieved by the cleavage of the dimeric complex 58 (Figure [5]) in the presence of pyridine to afford the monomeric complex 28a. Subsequent insertion of the Th–H into the C=N moiety of pyridine gives the thorium amido intermediate 28c. In the presence of HBpin, a subsequent Th–N/H–B σ-bond metathesis via the transition state 28d affords the final hydroborated product 28e and concurrently regenerates the active species 28a to complete the cycle.

# 5
Conclusions and Outlook
In summary, we have extensively reviewed recent advances in organo-f-element complex mediated the hydroborations of alkenes, carbonyls, imines, and nitriles. Despite the highly oxophilic nature of the lanthanide and actinide metal centers, the complexes displayed high catalytic efficiencies and high selectivities throughout the transformations, even when using oxygen-containing borane reagents. Noteworthy to point out that in the process of the hydroboration of carbonyl compounds, regeneration of Ln–H or An–H active species from Ln–O or An–O bonds is achieved, refreshing our traditional understanding of the expected sluggish Ln–O or An–O bonds. Another reaction that deserves attention is the dearomatization of N-heteroaromatics compounds by organo-f-element complexes via hydroboration. Dearomatized N-heteroaromatics, such as 1,2-dihydropyridine, play a significant role in the pharmaceutical fields, and until now, only few catalytic systems were available to disrupt the aromaticity of N-heteroaromatics. Organo-f-complexes displayed their unique advantage in this process when considering their high 1,2-regioselectivity, high catalytic turnovers, and more importantly, high earth abundance.
Despite the advances presented in this review, a large number of challenging and important reactions are still untapped. For example, the development and utilization of C1 sources (CO2 and CO), which is a green and sustainable strategy to convert these gases into value-added C1 compounds, such as methanol, formic acid, etc. Many stoichiometric reactions between organo-f-complexes with CO2 or CO have been reported,[59] and some transition metals catalyze the hydroboration of CO2.[60] However, no reports on organo-f-complexes mediating the hydroboration of CO2 or CO in a catalytic way have been disclosed. We need to ask, how far we can advance in transforming oxygen-containing substrates using organo-f-complexes? Despite recent advances in transforming oxygen-containing substrates, such as alcohol,[6c] [d] [f] aldehyde,[43a] ester,[42,44] etc., how about more challenging substrates? Such as water? For example, catalytically making H2 from H2O; some uranium complexes have been reported to be feasible.[61] In general, organo-f-element complex mediated hydroboration is still in its early stage; expansion into other substrates and deeper understanding of the catalytic mechanism of this type reaction is still required.
#
#
-
References
- 1a Arnold PL, Turner ZR. Nat. Rev. Chem. 2017; 1: 0002
- 1b Arnold PL. Chem. Commun. 2011; 47: 9005
- 1c Fox AR, Bart SC, Meyer K, Cummins CC. Nature 2008; 455: 341
- 1d Karmel IS, Batrice RJ, Eisen MS. Inorganics 2015; 3: 392
- 1e Andrea T, Eisen MS. Chem. Soc. Rev. 2008; 37: 550
- 1f Liu H, Ghatak T, Eisen MS. Chem. Commun. 2017; 53: 11278
- 1g Hong SW, Tian S, Metz MV, Marks TJ. J. Am. Chem. Soc. 2003; 125: 14768
- 1h Yu X, Marks TJ. Organometallics 2007; 26: 365
- 1i Edelmann FT. Coord. Chem. Rev. 2018; 370: 129
- 2 Marks TJ. Organometallics 2013; 32: 1133
- 3a Li YW, Marks TJ. J. Am. Chem. Soc. 1998; 120: 1757
- 3b Li YW, Marks TJ. J. Am. Chem. Soc. 1996; 118: 9295
- 4a Hong S, Marks TJ. Acc. Chem. Res. 2004; 37: 673
- 4b Stubbert BD, Marks TJ. J. Am. Chem. Soc. 2007; 129: 4253
- 4c Haar CM, Stern CL, Marks TJ. Organometallics 1996; 15: 1765
- 5 Weiss CJ, Marks TJ. Dalton Trans. 2010; 39: 6576
- 6a Yu X, Seo S, Marks TJ. J. Am. Chem. Soc. 2007; 129: 7244
- 6b Nolan SP, Stern D, Marks TJ. J. Am. Chem. Soc. 1989; 111: 7844
- 6c Batrice RJ, Kefalidis CE, Maron L, Eisen MS. J. Am. Chem. Soc. 2016; 138: 2114
- 6d Liu H, Khononov M, Fridman N, Tamm M, Eisen MS. Inorg. Chem. 2017; 56: 3153
- 6e Ghatak T, Fridman N, Eisen MS. Organometallics 2017; 36: 1296
- 6f Wobser SD, Marks TJ. Organometallics 2013; 32: 2517
- 7a Suzuki A. Proc. Jpn. Acad., Ser. B 2004; 80: 359
- 7b King RB. Chem. Rev. 2001; 101: 1119
- 7c Piers WE. Adv. Organomet. Chem. 2005; 52: 1
- 7d Staubitz A, Robertson AP. M, Sloan ME, Manners I. Chem. Rev. 2010; 110: 4023
- 7e Curran DP, Solovyev A, Brahmi MM, Fensterbank L, Malacria M, Lacote E. Angew. Chem. Int. Ed. 2011; 50: 10294
- 8 Brown HC, Rao BC. S. J. Am. Chem. Soc. 1959; 81: 6423
- 9a Jaladi AK, Shin WK, An DK. RSC Adv. 2019; 9: 26483
- 9b Bisai MK, Yadav S, Das T, Vanka K, Sen SS. Chem. Commun. 2019; 55: 11711
- 9c Wang Z.-C, Wang M, Gao J, Shi S.-L, Xu Y. Org. Chem. Front. 2019; 6: 2949
- 9d Pollard VA, Orr SA, McLellan R, Kennedy AR, Hevia E, Mulvey RE. Chem. Commun. 2018; 54: 1233
- 9e Bisai MK, Das T, Vanka K, Sen SS. Chem. Commun. 2018; 54: 6843
- 9f Brown HC, Narasimhan S. Organometallics 1982; 1: 762
- 10a Liu T, He J, Zhang Y. Org. Chem. Front. 2019; 6: 2749
- 10b Harinath A, Bhattacharjee J, Nayek HP, Panda TK. Dalton Trans. 2018; 47: 12613
- 10c Osseili H, Mukherjee D, Spaniol TP, Okuda J. Chem. Eur. J. 2017; 23: 14292
- 10d Mukherjee D, Osseili H, Spaniol TP, Okuda J. J. Am. Chem. Soc. 2016; 138: 10790
- 11a Li Y, Wu M, Chen H, Xu D, Qu L, Zhang J, Bai R, Lan Y. Front. Chem. 2019; 7: 149 ; DOI: org/10.3389/fchem.2019.00149
- 11b Magre M, Maity B, Falconnet A, Cavallo L, Rueping M. Angew. Chem. Int. Ed. 2019; 58: 7025
- 11c Lawson JR, Wilkins LC, Melen RL. Chem. Eur. J. 2017; 23: 10997
- 11d Weetman C, Hill MS, Mahon MF. Polyhedron 2016; 103: 115
- 11e Weetman C, Hill MS, Mahon MF. Chem. Eur. J. 2016; 22: 7158
- 11f Weetman C, Anker MD, Arrowsmith M, Hill MS, Kociok-Köhn G, Liptrot DJ, Mahon MF. Chem. Sci. 2016; 7: 628
- 11g Mukherjee D, Shirase S, Spaniol TP, Mashima K, Okuda J. Chem. Commun. 2016; 52: 13155
- 11h Weetman C, Hill MS, Mahon MF. Chem. Commun. 2015; 51: 14477
- 11i Mukherjee D, Ellern A, Sadow AD. Chem. Sci. 2014; 5: 959
- 11j Arrowsmith M, Hill MS, Kociok-Köhn G. Chem. Eur. J. 2013; 19: 2776
- 11k Arrowsmith M, Hadlington TJ, Hill MS, Kociok-Köhn G. Chem. Commun. 2012; 48: 4567
- 11l Arrowsmith M, Hill MS, Hadlington T, Kociok-Köhn G, Weetman C. Organometallics 2011; 30: 5556
- 11m Ingleson MF, Barrio JP, Bacsa J, Steiner A, Darling GR, Jones JT. A, Khimyak YZ, Rosseinsky MJ. Angew. Chem. Int. Ed. 2009; 48: 2012
- 12a Shen Q, Ma X, Li W, Liu W, Ding Y, Yang Z, Roesky HW. Chem. Eur. J. 2019; 25: 11918
- 12b Ding Y, Ma X, Liu Y, Liu W, Yang Z, Roesky HW. Organometallics 2019; 38: 3092
- 12c Liu W, Ding Y, Jin D, Shen Q, Yan B, Ma X, Yang Z. Green Chem. 2019; 21: 3812
- 12d Pollard VA, Fuentes MA, Kennedy AR, McLellan R, Mulvey RE. Angew. Chem. Int. Ed. 2018; 57: 10651
- 12e Bismuto A, Cowley MJ, Thomas SP. ACS Catal. 2018; 8: 2001
- 12f Gorgas N, Alves LG, Stoeger B, Martins AM, Veiros LF, Kirchner K. J. Am. Chem. Soc. 2017; 139: 8130
- 12g Jakhar VK, Barman MK, Nembenna S. Org. Lett. 2016; 18: 4710
- 12h Bismuto A, Thomas SP, Cowley MJ. Angew. Chem. Int. Ed. 2016; 55: 15356
- 13a Mandal S, Mandal S, Geetharani K. Chem. Asian J. 2019; DOI: in press; org/10.1002/asia.201900839.
- 13b Procter RJ, Uzelac M, Cid J, Rushworth PJ, Ingleson MJ. ACS Catal. 2019; 9: 5760
- 13c Procter RJ, Uzelac M, Cid J, Rushworth PJ, Ingleson MJ. ACS Catal. 2019; 9: 5760
- 13d Lortie JL, Dudding T, Gabidullin BM, Nikonov GI. ACS Catal. 2017; 7: 8454
- 14a Khusainova LI, Khafizova LO, Ryazanov KS, Tyumkina TV, Dzhemilev UM. J. Organomet. Chem. 2019; 898: 120858
- 14b Harinath A, Bhattcharjee J, Gorantla KR, Mallik BS, Panda TK. Eur. J. Org. Chem. 2018; 2018: 3180
- 14c Oluyadi AA, Ma S, Muhoro CN. Organometallics 2013; 32: 70
- 14d Liu D, Lin ZY. Organometallics 2002; 21: 4750
- 14e He XM, Hartwig JF. J. Am. Chem. Soc. 1996; 118: 1696
- 15a Ghosh C, Kim S, Mena MR, Kim J.-H, Pal R, Rock CL, Groy TL, Baik M.-H, Trovitch RJ. J. Am. Chem. Soc. 2019; 141: 15327
- 15b Zhang G, Li S, Wu J, Zeng H, Mo Z, Davis K, Zheng S. Org. Chem. Front. 2019; 6: 3228
- 15c Duvvuri K, Dewese KR, Parsutkar MM, Jing SM, Mehta MM, Gallucci JC, RajanBabu TV. J. Am. Chem. Soc. 2019; 141: 7365
- 15d Zhang L, Zuo Z, Wan X, Huang Z. J. Am. Chem. Soc. 2014; 136: 15501
- 15e Chen X, Cheng Z, Lu Z. ACS Catal. 2019; 9: 4025
- 16a Vijaykumar G, Bhunia M, Mandal SK. Dalton Trans. 2019; 48: 5779
- 16b Tamang SR, Singh A, Unruh DK, Findlater M. ACS Catal. 2018; 8: 6186
- 16c Kamei T, Nishino S, Shimada T. Tetrahedron Lett. 2018; 59: 2896
- 16d Nakamura G, Nakajima Y, Matsumoto K, Srinivas V, Shimada S. Catal. Sci. Technol. 2017; 7: 3196
- 16e Liu T, Meng W, Ma Q.-Q, Zhang J, Li H, Li S, Zhao Q, Chen X. Dalton Trans. 2017; 46: 4504
- 16f Li J.-F, Wei Z.-Z, Wang Y.-Q, Ye M. Green Chem. 2017; 19: 4498
- 16g Touney EE, Van Hoveln R, Buttke CT, Freidberg MD, Guzei IA, Schomaker JM. Organometallics 2016; 35: 3436
- 16h Ely RJ, Morken JP. J. Am. Chem. Soc. 2010; 132: 2534
- 17a Chen J.-Y, Liao R.-Z. Organometallics 2019; 38: 3267
- 17b Cruz TF. C, Pereira LC. J, Waerenborgh JC, Veiros LF, Gomes PT. Catal. Sci. Technol. 2019; 9: 3347
- 17c Chen J, Xi T, Lu Z. Org. Lett. 2014; 16: 6452
- 17d Zhang L, Peng D, Leng X, Huang Z. Angew. Chem. Int. Ed. 2013; 52: 3676
- 17e Obligacion JV, Chirik PJ. Org. Lett. 2013; 15: 2680
- 17f Greenhalgh MD, Thomas SP. Chem. Commun. 2013; 49: 11230
- 17g Wu JY, Moreau B, Ritter T. J. Am. Chem. Soc. 2009; 131: 12915
- 18a Huang J, Yan W, Tan C, Wu W, Jiang H. Chem. Commun. 2018; 54: 1770
- 18b Zhu C, Yang B, Qiu Y, Backvall J.-E. Chem. Eur. J. 2016; 22: 2939
- 18c Matsumoto Y, Naito M, Hayashi T. Organometallics 1992; 11: 2732
- 19a DiBenedetto TA, Parsons AM, Jones WD. Organometallics 2019; 38: 3322
- 19b Jang WJ, Kang B.-N, Lee JH, Choi YM, Kim C.-H, Yun J. Org. Biomol. Chem. 2019; 17: 5249
- 19c Armstrong MK, Lalic G. J. Am. Chem. Soc. 2019; 141: 6173
- 19d Jang WJ, Lee WL, Moon JH, Lee JY, Yun J. Org. Lett. 2016; 18: 1390
- 19e Semba K, Shinomiya M, Fujihara T, Terao J, Tsuji Y. Chem. Eur. J. 2013; 19: 7125
- 19f Feng X, Jeon H, Yun J. Angew. Chem. Int. Ed. 2013; 52: 3989
- 19g Noh D, Chea H, Ju J, Yun J. Angew. Chem. Int. Ed. 2009; 48: 6062
- 20a Kim HT, Ha H, Kang G, Kim OS, Ryu H, Biswas AK, Lim SM, Baik M.-H, Joo JM. Angew. Chem. Int. Ed. 2017; 56: 16262
- 20b Dietz M, Johnson A, Martínez-Martínez A, Weller AS. Inorg. Chim. Acta 2019; 491: 9
- 20c Lata CJ, Crudden CM. J. Am. Chem. Soc. 2010; 132: 131
- 20d Carroll AM, O’Sullivan TP, Guiry PJ. Adv. Synth. Catal. 2005; 347: 609
- 20e Rubina M, Rubin M, Gevorgyan V. J. Am. Chem. Soc. 2003; 125: 7198
- 20f Kwong FY, Yang QC, Mak TC. W, Chan AS. C, Chan KS. J. Org. Chem. 2002; 67: 2769
- 20g Mannig D, Noth H. Angew. Chem. Int. Ed. 1985; 24: 878
- 21a Wang G, Liang X, Chen L, Gao Q, Wang J.-G, Zhang P, Peng Q, Xu S. Angew. Chem. Int. Ed. 2019; 58: 8187
- 21b Yamamoto Y, Fujikawa R, Umemoto T, Miyaura N. Tetrahedron 2004; 60: 10695
- 21c Ohmura T, Yamamoto Y, Miyaura N. J. Am. Chem. Soc. 2000; 122: 4990
- 22a Wang Y, Guan R, Sivaguru P, Cong X, Bi X. Org. Lett. 2019; 21: 4035
- 22b Yoshida H, Kageyuki I, Takaki K. Org. Lett. 2014; 16: 3512
- 23a Li T, Zhang J, Cui C. Chin. J. Chem. 2019; 37: 679
- 23b Taniguchi T, Curran DP. Angew. Chem. Int. Ed. 2014; 53: 13150
- 24 Xu X, Yan D, Zhu Z, Kang Z, Yao Y, Shen Q, Xue M. ACS Omega 2019; 4: 6775
- 25a Chong CC, Kinjo R. ACS Catal. 2015; 5: 3238
- 25b Shegavi ML, Bose SK. Catal. Sci. Technol. 2019; 9: 3307
- 25c Tamang SR, Findlater M. Molecules 2019; 24: 3194
- 25d Crudden CM, Edwards D. Eur. J. Org. Chem. 2003; 2003: 4695
- 26 Harrison KN, Marks TJ. J. Am. Chem. Soc. 1992; 114: 9220
- 27a Gagne MR, Stern CL, Marks TJ. J. Am. Chem. Soc. 1992; 114: 275
- 27b Jeske G, Lauke H, Mauermann H, Swepston PN, Schumann H, Marks TJ. J. Am. Chem. Soc. 1985; 107: 8091
- 28 Evans DA, Muci AR, Stuermer R. J. Org. Chem. 1993; 58: 5307
- 29 Bijpost EA, Duchateau R, Teuben JH. J. Mol. Catal. A: Chem. 1995; 95: 121
- 30 Horino Y, Livinghouse T, Stan M. Synlett 2004; 2639
- 31 Villiers C, Ephritikhine M. J. Chem. Soc., Chem. Commun. 1995; 979
- 32 Molander GA, Pfeiffer D. Org. Lett. 2001; 3: 361
- 33 Kulkarni SA, Koga N. J. Mol. Struct.: THEOCHEM 1999; 461–462: 297
- 34a Manna K, Ji P, Greene FX, Lin W. J. Am. Chem. Soc. 2016; 138: 7488
- 34b Eedugurala N, Wang Z, Chaudhary U, Nelson N, Kandel K, Kobayashi T, Slowing II, Pruski M, Sadow AD. ACS Catal. 2015; 5: 7399
- 34c Kaithal A, Chatterjee B, Gunanathan C. Org. Lett. 2015; 17: 4790
- 34d Hadlington TJ, Hermann M, Frenking G, Jones C. J. Am. Chem. Soc. 2014; 136: 3028
- 34e Chong CC, Hirao H, Kinjo R. Angew. Chem. Int. Ed. 2015; 54: 190
- 35 Weidner VL, Barger CJ, Delferro M, Lohr TL, Marks TJ. ACS Catal. 2017; 7: 1244
- 36 Xue M, Wu Z, Hong Y, Shen Q. WO 2018000401, 2018
- 37a Chen S, Yan D, Xue M, Hong Y, Yao Y, Shen Q. Org. Lett. 2017; 19: 3382
- 37b Yan D, Dai P, Chen S, Xue M, Yao Y, Shen Q, Bao X. Org. Biomol. Chem. 2018; 16: 2787
- 38a Xue M, Hong Y, Chen S, Shen Q. WO 2018000400, 2018
- 38b Xue M, Zhu Z, Hong Y, Shen Q, Zheng Y. WO 2018000402, 2018
- 39 Wang W, Shen X, Zhao F, Jiang H, Yao W, Pullarkat SA, Xu L, Ma M. J. Org. Chem. 2018; 83: 69
- 40 Zhu Z, Dai P, Wu Z, Xue M, Yao Y, Shen Q, Bao X. Catal. Commun. 2018; 112: 26
- 41a Giffels G, Dreisbach C, Kragl U, Weigerding M, Waldmann H, Wandrey C. Angew. Chem. Int. Ed. 1995; 34: 2005
- 41b Molvinger K, Lopez M, Court J. Tetrahedron Lett. 1999; 40: 8375
- 41c Blake AJ, Cunningham A, Ford A, Teat SJ, Woodward S. Chem. Eur. J. 2000; 6: 3586
- 41d Fu IP, Uang B.-J. Tetrahedron: Asymmetry 2001; 12: 45
- 41e Song P, Lu C, Fei Z, Zhao B, Yao Y. J. Org. Chem. 2018; 83: 6093
- 42 Barger CJ, Motta A, Weidner VL, Lohr TL, Marks TJ. ACS Catal. 2019; 9: 9015
- 43a Karmel IS. R, Fridman N, Tamm M, Eisen MS. J. Am. Chem. Soc. 2014; 136: 17180
- 43b Karmel IS. R, Fridman N, Tamm M, Eisen MS. Organometallics 2015; 34: 2933
- 43c Liu H, Khononov M, Fridman N, Tamm M, Eisen MS. J. Organomet. Chem. 2017; 857: 123
- 43d Ghatak T, Drucker S, Fridman N, Eisen MS. Dalton Trans. 2017; 46: 12005
- 43e Liu H, Eisen MS. Organometallics 2017; 36: 1461
- 44 Karmel IS. R, Khononov M, Tamm M, Eisen MS. Catal. Sci. Technol. 2015; 5: 5110
- 45 Liu H, Fridman N, Tamm M, Eisen MS. Organometallics 2017; 36: 3896
- 46 Ghatak T, Makarov K, Fridman N, Eisen MS. Chem. Commun. 2018; 54: 11001
- 47a Xie J.-H, Zhu S.-F, Zhou Q.-L. Chem. Rev. 2011; 111: 1713
- 47b Werkmeister S, Junge K, Beller M. Org. Process Res. Dev. 2014; 18: 289
- 47c Bornschein C, Werkmeister S, Wendt B, Jiao H, Alberico E, Baumann W, Junge H, Junge K, Beller M. Nat. Commun. 2014; 5: 4111
- 47d Yap AJ, Masters AF, Maschmeyer T. ChemCatChem 2012; 4: 1179
- 47e Segobia DJ, Trasarti AF, Apesteguía CR. Appl. Catal., A 2012; 445–446: 69
- 47f Li Y, Gong Y, Xu X, Zhang P, Li H, Wang Y. Catal. Commun. 2012; 28: 9
- 47g Rajesh K, Dudle B, Blacque O, Berke H. Adv. Synth. Catal. 2011; 353: 1479
- 47h Chatterjee M, Kawanami H, Sato M, Ishizaka T, Yokoyama T, Suzuki T. Green Chem. 2010; 12: 87
- 47i Haddenham D, Pasumansky L, DeSoto J, Eagon S, Singaram B. J. Org. Chem. 2009; 74: 1964
- 47j Mukherjee A, Srimani D, Chakraborty S, Ben-David Y, Milstein D. J. Am. Chem. Soc. 2015; 137: 8888
- 47k Chakraborty S, Leitus G, Milstein D. Chem. Commun. 2016; 52: 1812
- 48a Colyer JT, Andersen NG, Tedrow JS, Soukup TS, Faul MM. J. Org. Chem. 2006; 71: 6859
- 48b Dorsey AD, Barbarow JE, Trauner D. Org. Lett. 2003; 5: 3237
- 48c Mollet K, D’hooghe M, De Kimpe N. J. Org. Chem. 2011; 76: 264
- 49a Wu J, Zeng H, Cheng J, Zheng S, Golen JA, Manke DR, Zhang G. J. Org. Chem. 2018; 83: 9442
- 49b Ben-Daat H, Rock CL, Flores M, Groy TL, Bowman AC, Trovitch RJ. Chem. Commun. 2017; 53: 7333
- 49c Ibrahim AD, Entsminger SW, Fout AR. ACS Catal. 2017; 7: 3730
- 50 Kaithal A, Chatterjee B, Gunanathan C. J. Org. Chem. 2016; 81: 11153
- 51a Khalimon AY, Farha P, Kuzmina LG, Nikonov GI. Chem. Commun. 2012; 48: 455
- 51b Fernandes AC, Romão CC. Tetrahedron Lett. 2007; 48: 9176
- 51c Hayes KS. Appl. Catal., A 2001; 221: 187
- 52 Baker RT, Calabrese JC, Westcott SA. J. Organomet. Chem. 1995; 498: 109
- 53 Dudnik AS, Weidner VL, Motta A, Delferro M, Marks TJ. Nat. Chem. 2014; 6: 1100
- 54a Oshima K, Ohmura T, Suginome M. J. Am. Chem. Soc. 2012; 134: 3699
- 54b Park S, Chang S. Angew. Chem. Int. Ed. 2017; 56: 7720
- 55 Huang Z, Wang S, Zhu X, Yuan Q, Wei Y, Zhou S, Mu X. Inorg. Chem. 2018; 57: 15069
- 56 Saha S, Eisen MS. ACS Catal. 2019; 9: 5947
- 57 Liu H, Kulbitski K, Tamm M, Eisen MS. Chem. Eur. J. 2018; 24: 5738
- 58 Liu H, Khononov M, Eisen MS. ACS Catal. 2018; 8: 3673
- 59a Manriquez JM, Fagan PJ, Marks TJ, Day CS, Day VW. J. Am. Chem. Soc. 1978; 100: 7112
- 59b Moloy KG, Marks TJ. Inorg. Chim. Acta 1985; 110: 127
- 59c Seo S, Yu X, Marks TJ. J. Am. Chem. Soc. 2009; 131: 263
- 59d Cooper O, Camp C, Pécaut J, Kefalidis CE, Maron L, Gambarelli S, Mazzanti M. J. Am. Chem. Soc. 2014; 136: 6716
- 60a Shintani R, Nozaki K. Organometallics 2013; 32: 2459
- 60b Zhang L, Cheng J, Hou Z. Chem. Commun. 2013; 49: 4782
- 61 Halter DP, Heinemann FW, Maron L, Meyer K. Nat. Chem. 2018; 10: 259
-
References
- 1a Arnold PL, Turner ZR. Nat. Rev. Chem. 2017; 1: 0002
- 1b Arnold PL. Chem. Commun. 2011; 47: 9005
- 1c Fox AR, Bart SC, Meyer K, Cummins CC. Nature 2008; 455: 341
- 1d Karmel IS, Batrice RJ, Eisen MS. Inorganics 2015; 3: 392
- 1e Andrea T, Eisen MS. Chem. Soc. Rev. 2008; 37: 550
- 1f Liu H, Ghatak T, Eisen MS. Chem. Commun. 2017; 53: 11278
- 1g Hong SW, Tian S, Metz MV, Marks TJ. J. Am. Chem. Soc. 2003; 125: 14768
- 1h Yu X, Marks TJ. Organometallics 2007; 26: 365
- 1i Edelmann FT. Coord. Chem. Rev. 2018; 370: 129
- 2 Marks TJ. Organometallics 2013; 32: 1133
- 3a Li YW, Marks TJ. J. Am. Chem. Soc. 1998; 120: 1757
- 3b Li YW, Marks TJ. J. Am. Chem. Soc. 1996; 118: 9295
- 4a Hong S, Marks TJ. Acc. Chem. Res. 2004; 37: 673
- 4b Stubbert BD, Marks TJ. J. Am. Chem. Soc. 2007; 129: 4253
- 4c Haar CM, Stern CL, Marks TJ. Organometallics 1996; 15: 1765
- 5 Weiss CJ, Marks TJ. Dalton Trans. 2010; 39: 6576
- 6a Yu X, Seo S, Marks TJ. J. Am. Chem. Soc. 2007; 129: 7244
- 6b Nolan SP, Stern D, Marks TJ. J. Am. Chem. Soc. 1989; 111: 7844
- 6c Batrice RJ, Kefalidis CE, Maron L, Eisen MS. J. Am. Chem. Soc. 2016; 138: 2114
- 6d Liu H, Khononov M, Fridman N, Tamm M, Eisen MS. Inorg. Chem. 2017; 56: 3153
- 6e Ghatak T, Fridman N, Eisen MS. Organometallics 2017; 36: 1296
- 6f Wobser SD, Marks TJ. Organometallics 2013; 32: 2517
- 7a Suzuki A. Proc. Jpn. Acad., Ser. B 2004; 80: 359
- 7b King RB. Chem. Rev. 2001; 101: 1119
- 7c Piers WE. Adv. Organomet. Chem. 2005; 52: 1
- 7d Staubitz A, Robertson AP. M, Sloan ME, Manners I. Chem. Rev. 2010; 110: 4023
- 7e Curran DP, Solovyev A, Brahmi MM, Fensterbank L, Malacria M, Lacote E. Angew. Chem. Int. Ed. 2011; 50: 10294
- 8 Brown HC, Rao BC. S. J. Am. Chem. Soc. 1959; 81: 6423
- 9a Jaladi AK, Shin WK, An DK. RSC Adv. 2019; 9: 26483
- 9b Bisai MK, Yadav S, Das T, Vanka K, Sen SS. Chem. Commun. 2019; 55: 11711
- 9c Wang Z.-C, Wang M, Gao J, Shi S.-L, Xu Y. Org. Chem. Front. 2019; 6: 2949
- 9d Pollard VA, Orr SA, McLellan R, Kennedy AR, Hevia E, Mulvey RE. Chem. Commun. 2018; 54: 1233
- 9e Bisai MK, Das T, Vanka K, Sen SS. Chem. Commun. 2018; 54: 6843
- 9f Brown HC, Narasimhan S. Organometallics 1982; 1: 762
- 10a Liu T, He J, Zhang Y. Org. Chem. Front. 2019; 6: 2749
- 10b Harinath A, Bhattacharjee J, Nayek HP, Panda TK. Dalton Trans. 2018; 47: 12613
- 10c Osseili H, Mukherjee D, Spaniol TP, Okuda J. Chem. Eur. J. 2017; 23: 14292
- 10d Mukherjee D, Osseili H, Spaniol TP, Okuda J. J. Am. Chem. Soc. 2016; 138: 10790
- 11a Li Y, Wu M, Chen H, Xu D, Qu L, Zhang J, Bai R, Lan Y. Front. Chem. 2019; 7: 149 ; DOI: org/10.3389/fchem.2019.00149
- 11b Magre M, Maity B, Falconnet A, Cavallo L, Rueping M. Angew. Chem. Int. Ed. 2019; 58: 7025
- 11c Lawson JR, Wilkins LC, Melen RL. Chem. Eur. J. 2017; 23: 10997
- 11d Weetman C, Hill MS, Mahon MF. Polyhedron 2016; 103: 115
- 11e Weetman C, Hill MS, Mahon MF. Chem. Eur. J. 2016; 22: 7158
- 11f Weetman C, Anker MD, Arrowsmith M, Hill MS, Kociok-Köhn G, Liptrot DJ, Mahon MF. Chem. Sci. 2016; 7: 628
- 11g Mukherjee D, Shirase S, Spaniol TP, Mashima K, Okuda J. Chem. Commun. 2016; 52: 13155
- 11h Weetman C, Hill MS, Mahon MF. Chem. Commun. 2015; 51: 14477
- 11i Mukherjee D, Ellern A, Sadow AD. Chem. Sci. 2014; 5: 959
- 11j Arrowsmith M, Hill MS, Kociok-Köhn G. Chem. Eur. J. 2013; 19: 2776
- 11k Arrowsmith M, Hadlington TJ, Hill MS, Kociok-Köhn G. Chem. Commun. 2012; 48: 4567
- 11l Arrowsmith M, Hill MS, Hadlington T, Kociok-Köhn G, Weetman C. Organometallics 2011; 30: 5556
- 11m Ingleson MF, Barrio JP, Bacsa J, Steiner A, Darling GR, Jones JT. A, Khimyak YZ, Rosseinsky MJ. Angew. Chem. Int. Ed. 2009; 48: 2012
- 12a Shen Q, Ma X, Li W, Liu W, Ding Y, Yang Z, Roesky HW. Chem. Eur. J. 2019; 25: 11918
- 12b Ding Y, Ma X, Liu Y, Liu W, Yang Z, Roesky HW. Organometallics 2019; 38: 3092
- 12c Liu W, Ding Y, Jin D, Shen Q, Yan B, Ma X, Yang Z. Green Chem. 2019; 21: 3812
- 12d Pollard VA, Fuentes MA, Kennedy AR, McLellan R, Mulvey RE. Angew. Chem. Int. Ed. 2018; 57: 10651
- 12e Bismuto A, Cowley MJ, Thomas SP. ACS Catal. 2018; 8: 2001
- 12f Gorgas N, Alves LG, Stoeger B, Martins AM, Veiros LF, Kirchner K. J. Am. Chem. Soc. 2017; 139: 8130
- 12g Jakhar VK, Barman MK, Nembenna S. Org. Lett. 2016; 18: 4710
- 12h Bismuto A, Thomas SP, Cowley MJ. Angew. Chem. Int. Ed. 2016; 55: 15356
- 13a Mandal S, Mandal S, Geetharani K. Chem. Asian J. 2019; DOI: in press; org/10.1002/asia.201900839.
- 13b Procter RJ, Uzelac M, Cid J, Rushworth PJ, Ingleson MJ. ACS Catal. 2019; 9: 5760
- 13c Procter RJ, Uzelac M, Cid J, Rushworth PJ, Ingleson MJ. ACS Catal. 2019; 9: 5760
- 13d Lortie JL, Dudding T, Gabidullin BM, Nikonov GI. ACS Catal. 2017; 7: 8454
- 14a Khusainova LI, Khafizova LO, Ryazanov KS, Tyumkina TV, Dzhemilev UM. J. Organomet. Chem. 2019; 898: 120858
- 14b Harinath A, Bhattcharjee J, Gorantla KR, Mallik BS, Panda TK. Eur. J. Org. Chem. 2018; 2018: 3180
- 14c Oluyadi AA, Ma S, Muhoro CN. Organometallics 2013; 32: 70
- 14d Liu D, Lin ZY. Organometallics 2002; 21: 4750
- 14e He XM, Hartwig JF. J. Am. Chem. Soc. 1996; 118: 1696
- 15a Ghosh C, Kim S, Mena MR, Kim J.-H, Pal R, Rock CL, Groy TL, Baik M.-H, Trovitch RJ. J. Am. Chem. Soc. 2019; 141: 15327
- 15b Zhang G, Li S, Wu J, Zeng H, Mo Z, Davis K, Zheng S. Org. Chem. Front. 2019; 6: 3228
- 15c Duvvuri K, Dewese KR, Parsutkar MM, Jing SM, Mehta MM, Gallucci JC, RajanBabu TV. J. Am. Chem. Soc. 2019; 141: 7365
- 15d Zhang L, Zuo Z, Wan X, Huang Z. J. Am. Chem. Soc. 2014; 136: 15501
- 15e Chen X, Cheng Z, Lu Z. ACS Catal. 2019; 9: 4025
- 16a Vijaykumar G, Bhunia M, Mandal SK. Dalton Trans. 2019; 48: 5779
- 16b Tamang SR, Singh A, Unruh DK, Findlater M. ACS Catal. 2018; 8: 6186
- 16c Kamei T, Nishino S, Shimada T. Tetrahedron Lett. 2018; 59: 2896
- 16d Nakamura G, Nakajima Y, Matsumoto K, Srinivas V, Shimada S. Catal. Sci. Technol. 2017; 7: 3196
- 16e Liu T, Meng W, Ma Q.-Q, Zhang J, Li H, Li S, Zhao Q, Chen X. Dalton Trans. 2017; 46: 4504
- 16f Li J.-F, Wei Z.-Z, Wang Y.-Q, Ye M. Green Chem. 2017; 19: 4498
- 16g Touney EE, Van Hoveln R, Buttke CT, Freidberg MD, Guzei IA, Schomaker JM. Organometallics 2016; 35: 3436
- 16h Ely RJ, Morken JP. J. Am. Chem. Soc. 2010; 132: 2534
- 17a Chen J.-Y, Liao R.-Z. Organometallics 2019; 38: 3267
- 17b Cruz TF. C, Pereira LC. J, Waerenborgh JC, Veiros LF, Gomes PT. Catal. Sci. Technol. 2019; 9: 3347
- 17c Chen J, Xi T, Lu Z. Org. Lett. 2014; 16: 6452
- 17d Zhang L, Peng D, Leng X, Huang Z. Angew. Chem. Int. Ed. 2013; 52: 3676
- 17e Obligacion JV, Chirik PJ. Org. Lett. 2013; 15: 2680
- 17f Greenhalgh MD, Thomas SP. Chem. Commun. 2013; 49: 11230
- 17g Wu JY, Moreau B, Ritter T. J. Am. Chem. Soc. 2009; 131: 12915
- 18a Huang J, Yan W, Tan C, Wu W, Jiang H. Chem. Commun. 2018; 54: 1770
- 18b Zhu C, Yang B, Qiu Y, Backvall J.-E. Chem. Eur. J. 2016; 22: 2939
- 18c Matsumoto Y, Naito M, Hayashi T. Organometallics 1992; 11: 2732
- 19a DiBenedetto TA, Parsons AM, Jones WD. Organometallics 2019; 38: 3322
- 19b Jang WJ, Kang B.-N, Lee JH, Choi YM, Kim C.-H, Yun J. Org. Biomol. Chem. 2019; 17: 5249
- 19c Armstrong MK, Lalic G. J. Am. Chem. Soc. 2019; 141: 6173
- 19d Jang WJ, Lee WL, Moon JH, Lee JY, Yun J. Org. Lett. 2016; 18: 1390
- 19e Semba K, Shinomiya M, Fujihara T, Terao J, Tsuji Y. Chem. Eur. J. 2013; 19: 7125
- 19f Feng X, Jeon H, Yun J. Angew. Chem. Int. Ed. 2013; 52: 3989
- 19g Noh D, Chea H, Ju J, Yun J. Angew. Chem. Int. Ed. 2009; 48: 6062
- 20a Kim HT, Ha H, Kang G, Kim OS, Ryu H, Biswas AK, Lim SM, Baik M.-H, Joo JM. Angew. Chem. Int. Ed. 2017; 56: 16262
- 20b Dietz M, Johnson A, Martínez-Martínez A, Weller AS. Inorg. Chim. Acta 2019; 491: 9
- 20c Lata CJ, Crudden CM. J. Am. Chem. Soc. 2010; 132: 131
- 20d Carroll AM, O’Sullivan TP, Guiry PJ. Adv. Synth. Catal. 2005; 347: 609
- 20e Rubina M, Rubin M, Gevorgyan V. J. Am. Chem. Soc. 2003; 125: 7198
- 20f Kwong FY, Yang QC, Mak TC. W, Chan AS. C, Chan KS. J. Org. Chem. 2002; 67: 2769
- 20g Mannig D, Noth H. Angew. Chem. Int. Ed. 1985; 24: 878
- 21a Wang G, Liang X, Chen L, Gao Q, Wang J.-G, Zhang P, Peng Q, Xu S. Angew. Chem. Int. Ed. 2019; 58: 8187
- 21b Yamamoto Y, Fujikawa R, Umemoto T, Miyaura N. Tetrahedron 2004; 60: 10695
- 21c Ohmura T, Yamamoto Y, Miyaura N. J. Am. Chem. Soc. 2000; 122: 4990
- 22a Wang Y, Guan R, Sivaguru P, Cong X, Bi X. Org. Lett. 2019; 21: 4035
- 22b Yoshida H, Kageyuki I, Takaki K. Org. Lett. 2014; 16: 3512
- 23a Li T, Zhang J, Cui C. Chin. J. Chem. 2019; 37: 679
- 23b Taniguchi T, Curran DP. Angew. Chem. Int. Ed. 2014; 53: 13150
- 24 Xu X, Yan D, Zhu Z, Kang Z, Yao Y, Shen Q, Xue M. ACS Omega 2019; 4: 6775
- 25a Chong CC, Kinjo R. ACS Catal. 2015; 5: 3238
- 25b Shegavi ML, Bose SK. Catal. Sci. Technol. 2019; 9: 3307
- 25c Tamang SR, Findlater M. Molecules 2019; 24: 3194
- 25d Crudden CM, Edwards D. Eur. J. Org. Chem. 2003; 2003: 4695
- 26 Harrison KN, Marks TJ. J. Am. Chem. Soc. 1992; 114: 9220
- 27a Gagne MR, Stern CL, Marks TJ. J. Am. Chem. Soc. 1992; 114: 275
- 27b Jeske G, Lauke H, Mauermann H, Swepston PN, Schumann H, Marks TJ. J. Am. Chem. Soc. 1985; 107: 8091
- 28 Evans DA, Muci AR, Stuermer R. J. Org. Chem. 1993; 58: 5307
- 29 Bijpost EA, Duchateau R, Teuben JH. J. Mol. Catal. A: Chem. 1995; 95: 121
- 30 Horino Y, Livinghouse T, Stan M. Synlett 2004; 2639
- 31 Villiers C, Ephritikhine M. J. Chem. Soc., Chem. Commun. 1995; 979
- 32 Molander GA, Pfeiffer D. Org. Lett. 2001; 3: 361
- 33 Kulkarni SA, Koga N. J. Mol. Struct.: THEOCHEM 1999; 461–462: 297
- 34a Manna K, Ji P, Greene FX, Lin W. J. Am. Chem. Soc. 2016; 138: 7488
- 34b Eedugurala N, Wang Z, Chaudhary U, Nelson N, Kandel K, Kobayashi T, Slowing II, Pruski M, Sadow AD. ACS Catal. 2015; 5: 7399
- 34c Kaithal A, Chatterjee B, Gunanathan C. Org. Lett. 2015; 17: 4790
- 34d Hadlington TJ, Hermann M, Frenking G, Jones C. J. Am. Chem. Soc. 2014; 136: 3028
- 34e Chong CC, Hirao H, Kinjo R. Angew. Chem. Int. Ed. 2015; 54: 190
- 35 Weidner VL, Barger CJ, Delferro M, Lohr TL, Marks TJ. ACS Catal. 2017; 7: 1244
- 36 Xue M, Wu Z, Hong Y, Shen Q. WO 2018000401, 2018
- 37a Chen S, Yan D, Xue M, Hong Y, Yao Y, Shen Q. Org. Lett. 2017; 19: 3382
- 37b Yan D, Dai P, Chen S, Xue M, Yao Y, Shen Q, Bao X. Org. Biomol. Chem. 2018; 16: 2787
- 38a Xue M, Hong Y, Chen S, Shen Q. WO 2018000400, 2018
- 38b Xue M, Zhu Z, Hong Y, Shen Q, Zheng Y. WO 2018000402, 2018
- 39 Wang W, Shen X, Zhao F, Jiang H, Yao W, Pullarkat SA, Xu L, Ma M. J. Org. Chem. 2018; 83: 69
- 40 Zhu Z, Dai P, Wu Z, Xue M, Yao Y, Shen Q, Bao X. Catal. Commun. 2018; 112: 26
- 41a Giffels G, Dreisbach C, Kragl U, Weigerding M, Waldmann H, Wandrey C. Angew. Chem. Int. Ed. 1995; 34: 2005
- 41b Molvinger K, Lopez M, Court J. Tetrahedron Lett. 1999; 40: 8375
- 41c Blake AJ, Cunningham A, Ford A, Teat SJ, Woodward S. Chem. Eur. J. 2000; 6: 3586
- 41d Fu IP, Uang B.-J. Tetrahedron: Asymmetry 2001; 12: 45
- 41e Song P, Lu C, Fei Z, Zhao B, Yao Y. J. Org. Chem. 2018; 83: 6093
- 42 Barger CJ, Motta A, Weidner VL, Lohr TL, Marks TJ. ACS Catal. 2019; 9: 9015
- 43a Karmel IS. R, Fridman N, Tamm M, Eisen MS. J. Am. Chem. Soc. 2014; 136: 17180
- 43b Karmel IS. R, Fridman N, Tamm M, Eisen MS. Organometallics 2015; 34: 2933
- 43c Liu H, Khononov M, Fridman N, Tamm M, Eisen MS. J. Organomet. Chem. 2017; 857: 123
- 43d Ghatak T, Drucker S, Fridman N, Eisen MS. Dalton Trans. 2017; 46: 12005
- 43e Liu H, Eisen MS. Organometallics 2017; 36: 1461
- 44 Karmel IS. R, Khononov M, Tamm M, Eisen MS. Catal. Sci. Technol. 2015; 5: 5110
- 45 Liu H, Fridman N, Tamm M, Eisen MS. Organometallics 2017; 36: 3896
- 46 Ghatak T, Makarov K, Fridman N, Eisen MS. Chem. Commun. 2018; 54: 11001
- 47a Xie J.-H, Zhu S.-F, Zhou Q.-L. Chem. Rev. 2011; 111: 1713
- 47b Werkmeister S, Junge K, Beller M. Org. Process Res. Dev. 2014; 18: 289
- 47c Bornschein C, Werkmeister S, Wendt B, Jiao H, Alberico E, Baumann W, Junge H, Junge K, Beller M. Nat. Commun. 2014; 5: 4111
- 47d Yap AJ, Masters AF, Maschmeyer T. ChemCatChem 2012; 4: 1179
- 47e Segobia DJ, Trasarti AF, Apesteguía CR. Appl. Catal., A 2012; 445–446: 69
- 47f Li Y, Gong Y, Xu X, Zhang P, Li H, Wang Y. Catal. Commun. 2012; 28: 9
- 47g Rajesh K, Dudle B, Blacque O, Berke H. Adv. Synth. Catal. 2011; 353: 1479
- 47h Chatterjee M, Kawanami H, Sato M, Ishizaka T, Yokoyama T, Suzuki T. Green Chem. 2010; 12: 87
- 47i Haddenham D, Pasumansky L, DeSoto J, Eagon S, Singaram B. J. Org. Chem. 2009; 74: 1964
- 47j Mukherjee A, Srimani D, Chakraborty S, Ben-David Y, Milstein D. J. Am. Chem. Soc. 2015; 137: 8888
- 47k Chakraborty S, Leitus G, Milstein D. Chem. Commun. 2016; 52: 1812
- 48a Colyer JT, Andersen NG, Tedrow JS, Soukup TS, Faul MM. J. Org. Chem. 2006; 71: 6859
- 48b Dorsey AD, Barbarow JE, Trauner D. Org. Lett. 2003; 5: 3237
- 48c Mollet K, D’hooghe M, De Kimpe N. J. Org. Chem. 2011; 76: 264
- 49a Wu J, Zeng H, Cheng J, Zheng S, Golen JA, Manke DR, Zhang G. J. Org. Chem. 2018; 83: 9442
- 49b Ben-Daat H, Rock CL, Flores M, Groy TL, Bowman AC, Trovitch RJ. Chem. Commun. 2017; 53: 7333
- 49c Ibrahim AD, Entsminger SW, Fout AR. ACS Catal. 2017; 7: 3730
- 50 Kaithal A, Chatterjee B, Gunanathan C. J. Org. Chem. 2016; 81: 11153
- 51a Khalimon AY, Farha P, Kuzmina LG, Nikonov GI. Chem. Commun. 2012; 48: 455
- 51b Fernandes AC, Romão CC. Tetrahedron Lett. 2007; 48: 9176
- 51c Hayes KS. Appl. Catal., A 2001; 221: 187
- 52 Baker RT, Calabrese JC, Westcott SA. J. Organomet. Chem. 1995; 498: 109
- 53 Dudnik AS, Weidner VL, Motta A, Delferro M, Marks TJ. Nat. Chem. 2014; 6: 1100
- 54a Oshima K, Ohmura T, Suginome M. J. Am. Chem. Soc. 2012; 134: 3699
- 54b Park S, Chang S. Angew. Chem. Int. Ed. 2017; 56: 7720
- 55 Huang Z, Wang S, Zhu X, Yuan Q, Wei Y, Zhou S, Mu X. Inorg. Chem. 2018; 57: 15069
- 56 Saha S, Eisen MS. ACS Catal. 2019; 9: 5947
- 57 Liu H, Kulbitski K, Tamm M, Eisen MS. Chem. Eur. J. 2018; 24: 5738
- 58 Liu H, Khononov M, Eisen MS. ACS Catal. 2018; 8: 3673
- 59a Manriquez JM, Fagan PJ, Marks TJ, Day CS, Day VW. J. Am. Chem. Soc. 1978; 100: 7112
- 59b Moloy KG, Marks TJ. Inorg. Chim. Acta 1985; 110: 127
- 59c Seo S, Yu X, Marks TJ. J. Am. Chem. Soc. 2009; 131: 263
- 59d Cooper O, Camp C, Pécaut J, Kefalidis CE, Maron L, Gambarelli S, Mazzanti M. J. Am. Chem. Soc. 2014; 136: 6716
- 60a Shintani R, Nozaki K. Organometallics 2013; 32: 2459
- 60b Zhang L, Cheng J, Hou Z. Chem. Commun. 2013; 49: 4782
- 61 Halter DP, Heinemann FW, Maron L, Meyer K. Nat. Chem. 2018; 10: 259