Key words
Amaryllidaceae - narciclasine - alkaloids - inflammation - cancer
Amaryllidaceae Alkaloids
The natural compound narciclasine was named after the plant genus Narcissus (daffodil), which belongs to the Amaryllidaceae (amaryllis) family. The medical use
of different Narcissus species dates back to ancient times. Famous physicians of this period, such as Hippocrates
of Kos (4th century BC) or Pedanius Dioscorides (1st century AD), recommended narcissus
oil as a treatment against cancer, in particular uterine tumors [1]. This tradition
was perpetuated during medieval times, e.g. in France by Henri de Mondeville (14th
century AD) [1]. The pharmacologically most interesting secondary metabolites present in plants
of the Amaryllidaceae family are alkaloids. Their scientific evaluation started in
1877 with the isolation of lycorine ([Fig. 1]) [2], the prototypical and most widely spread representative of the Amaryllidaceae alkaloids.
Its chemical structure was published in 1956 [3]. The Amaryllidaceae family consists of 75 genera with about 1600 species [4], from which approximately 500 different alkaloids have been identified up to now.
Based on the underlying skeleton, these compounds can be classified into at least
nine different groups. Some authors even expanded this classification to 18 groups
[5], [6], [7]. The most important prototypes of the nine classes are norbelladine, lycorine, homolycorine,
haemanthamine, tazettine, montanine, galanthamine, crinine, and narciclasine ([Fig. 1]) [7], [8], [9], [10]. The Amaryllidaceae alkaloids have been found to exert a huge variety of pharmacological
properties, such as antiproliferative, antitumor/cytotoxic, acetylcholinesterase inhibitory,
analgesic, hypotensive, antibacterial, and antifungal activities [11]. The intensive research of the last decades, consisting of isolation, structure
elucidation, analysis of structure-activity relationship, total synthesis, and pharmacological
characterization, has shed light on their tremendous potential. In fact, in 2000/2001,
the Amaryllidaceae alkaloid galanthamine ([Fig. 1]), which was discovered in Galanthus woronowii (Caucasian snowdrop), was approved for the treatment of mild to moderate forms of
Alzheimerʼs disease in Europe and in the USA.
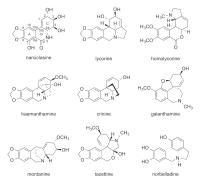 Fig. 1 Chemical structures of narciclasine and eight other prototypes of Amaryllidaceae
alkaloids.
Fig. 1 Chemical structures of narciclasine and eight other prototypes of Amaryllidaceae
alkaloids.
The Discovery of Narciclasine
The Discovery of Narciclasine
Narciclasine ([Fig. 1]) was first isolated in 1967 from bulbs of several Narcissus species within a research program looking for antigrowth factors [12]. One year later, a chemical structure was proposed [13], but turned out to be incorrect. It was revised in 1970 [14], [15] and the stereochemistry was fully elucidated by X-ray analysis in 1972 [16]. Narciclasine bears the substructures of isoquinoline, phenanthridine, and isocarbostyril.
Due to the amide structure, it is not a basic alkaloid. Interestingly, the Amaryllidaceae
alkaloid lycoricidinol, which was first isolated in 1968 as plant-growth regulator
from the bulbs of Lycoris radiata (red spider lily) [17], turned out to be identical to narciclasine [18]. Thus, narciclasine was discovered independently by two different groups at almost
the same time. In the following years, the compound as well as congeners thereof (e.g.
narciprimine, 7-deoxynarciclasine, trans-dihydronarciclasine, or 4-O-β-D-glucosylnarciclasine) were also found in a variety of other Amaryllidaceae genera
(e.g. Haemanthus, Galanthus, Hymenocallis, or Leucojum) [8], [19], [20], [21], [22], [23]. The amount of narciclasine in these plants was reported to vary from 1.5 mg/kg
(fresh bulbs) to 200 mg/kg [8], [19]. An intriguing discovery was made in 2005: narciclasine was found in the Texas grasshopper
Brachystola magna
[24]. As this animal does not prefer species of the Amaryllidaceae family as food, undiscovered
plant sources of narciclasine might still exist. Surprisingly, despite the intensive
research on narciclasine, the first total synthesis was not developed until 1997 [25]. Up to now, six different total syntheses of narciclasine and one of the enantiomeric
molecule have been published [26], [27]. The number of steps necessary to build up the compound ranges between 11 and 22.
The Biosynthesis of Narciclasine
The Biosynthesis of Narciclasine
Narciclasine originates from O-methylnorbelladine, the central precursor of all Amaryllidaceae alkaloids. The biosynthesis
of this precursor was intensively investigated in the late 1950s and early 1960s by
tracer experiments with radiolabeled precursors and intermediates [28]: O-methylnorbelladine derives from the aromatic amino acids phenylalanine and tyrosine.
Phenylalanine is transformed to protocatechuic aldehyde via trans-cinnamic acid, p-coumaric acid, and caffeic acid. Protocatechuic aldehyde reacts with tyramine, the
decarboxylated form of tyrosine, yielding an imine (Schiffʼs base) that is reduced
and methylated to O-methylnorbelladine. The subsequent biosynthetic route to narciclasine was studied
in the early 1970s [8], [19], [29], [30], [31], [32]: It was suggested to start with the cyclization of O-methylnorbelladine by a para-para phenol coupling reaction ([Fig. 2]). The resulting compound is eventually converted into narciclasine via the intermediate
11-hydroxyvittatine and by an elimination of two carbon atoms ([Fig. 2]). Although considerable progress has been made in the last years in the field of
metabolic engineering for the production of plant isoquinoline alkaloids [33], the knowledge about the biosynthesis of narciclasine has not expanded significantly
since the 1970s. Neither the involved enzymes nor their respective genes have been
characterized. This knowledge, however, would be strongly needed for a rational biotechnological
approach, in particular to solve the supply issue, by producing narciclasine in an
efficient way in plants or in microorganisms, e.g. by heterologous expression.
 Fig. 2 Proposed biosynthetic route from O-methylnorbelladine to narciclasine.
Fig. 2 Proposed biosynthetic route from O-methylnorbelladine to narciclasine.
Preclinical Knowledge About Narciclasine
Preclinical Knowledge About Narciclasine
The subsequently presented knowledge about the pharmacological actions, the underlying
mechanisms, and the target of narciclasine are graphically summarized in the [Fig. 3].
 Fig. 3 The most important biological actions, targets and mechanisms of narciclasine.
Fig. 3 The most important biological actions, targets and mechanisms of narciclasine.
Anti-tumor activity
Narciclasine inhibits cell growth by blocking protein biosynthesis. The initial publication describing the isolation of narciclasine also contains the
first evaluation of its bioactivity: The compound showed a strong mitosis-blocking
activity, since it effectively inhibited the growth of wheat grain radicles (0.05–0.5 µg/mL)
as well as of murine sarcoma cells in vivo (oral and subcutaneous application) [12]. The first study proposing a mechanistic explanation of these actions was published
in 1975: Carrasco et al. [34] reported that the alkaloid inhibits protein synthesis in rabbit reticulocytes as
well as in a yeast-derived cell-free system by blocking peptide bond formation at
the ribosome. These results were corroborated and largely expanded by further detailed
studies published between 1975 and 1978 by the group of Vazquez [35], [36], [37], [38], [39]: Narciclasine blocked protein synthesis with an IC50 of 70 nM and inhibited HeLa (human cervix carcinoma) cell growth with a comparable
IC50 of 100 nM. RNA synthesis was not affected, whereas DNA synthesis was slightly diminished,
but only at much higher concentrations. The mechanistic basis of this effect has not
been clarified, however, a direct interaction of narciclasine with the DNA (complex
formation) was excluded [40]. The binding site of narciclasine was located in the 60S subunit of the ribosome
and found to overlap with that of the known peptidyl transferase inhibitors anisomycin
and trichodermin. Two decades later, narciclasine was utilized as a chemical probe
to study the fine structure of the ribosomal peptidyl transferase center [41]. In 2014, X-ray crystallography was applied to decipher the exact binding mode of
16 different ribosome inhibitors, among them narciclasine, at the atomic level in
yeast ribosomes [42]. This study confirmed that narciclasine inhibits the step of peptide bond formation
during elongation by binding to the 60S tRNA A-site.
Mechanisms beyond ribosome inhibition. In the early 1990s, the group of G. Pettit reported about the broad cytotoxic activities
of narciclasine against a variety of cancer cells after the compound was tested in
the NCI panel of 60 human tumor cell lines [43]. The mean IC50 of narciclasine in this screening was 15.5 nM. Interestingly, melanoma cell lines
were the most sensitive tumor cells. Besides the in vitro screening approach, the NCI also performed a number of tests on tumor models in mice
[44]. However, the overall efficacy of narciclasine was only modest and associated with
a considerable toxicity. Surprisingly, the mechanisms underlying the narciclasine-induced
death of cancer cells was not investigated until 2007: Dumont et al. [45] showed that narciclasine was cytotoxic to all investigated cancer cell lines (IC50: 30 nM), whereas much higher concentrations were need to interfere with the viability
of fibroblasts (IC50: 7.5 µM). Ingrassia et al. [46] confirmed this selectivity towards cancer cells (mean IC50: 38 nM) and reported that endothelial cells (HUVECs) are more sensitive than fibroblasts,
since narciclasine inhibited the proliferation of endothelial cells with an IC50 of approx. 90 nM. This points towards a possible antiangiogenic action of the alkaloid.
In fact, unpublished data from our own lab confirm this hypothesis.
Narciclasine used at the high concentration of 1 µM was proven to cause apoptotic
cancer cell death via activation of the Fas and death receptor 4 (DR4) death-inducing
complex (DISC) and the subsequent recruitment of caspase-8 [45]. Interestingly, whether downstream effector caspases (e.g. caspase-3) are activated
directly or via mitochondria depends on the cell type: In the prostate cancer cell
line PC-3, narciclasine directly activated effector caspases via the Fas/DR4-triggered
assembly of DISC and the activation of caspase-8. In the breast cancer cell line MCF-7,
however, the activation of effector caspases depended on the processing of Bid, the
release of cytochrome c from mitochondria and the subsequent formation of the apoptosome
[45]. In human promyeloic HL-60 cells and in human oral cavity squamous carcinoma HSC-2
cells, also low concentrations of narciclasine (18 and 50 nM, respectively) have been
proven to induce apoptosis (cell shrinkage, DNA fragmentation, caspase-3 activation)
[47].
In contrast, narciclasine used at the concentration of 100 nM was also found to impair
tumor cell growth without inducing apoptosis: The alkaloid blocked the proliferation
and migration of glioblastoma cells in vitro, but did not trigger apoptotic cell death [48]. The authors of the study did not claim the inhibition of translation as mechanistic
basis of the antitumor effect, but provided evidence that narciclasine activates the
small GTPase RhoA [48]. Of note, RhoA was not suggested or investigated as a direct target of narciclasine.
The activation of RhoA eventually led to the formation of F-actin stress fibers via the Rho kinase/LIM kinase/cofilin pathway. The increased generation of stress fibers
was speculated to be the basis for the inhibition of cytokinesis and thus mitosis/proliferation,
as well as for the decreased migratory capacity of glioblastoma cells. In the same
study, the authors also tested narciclasine in vivo in a murine orthotopic model of human glioblastoma tumors. In contrast to the above
mentioned disappointing in vivo results gathered by the NCI [44], narciclasine (1 mg/kg, orally or via tail vein
injections) was able to considerably act against the glioblastoma tumor and to significantly
increase the survival of the glioblastoma-bearing mice [48].
Van Goietsenoven et al. [49] studied narciclasine in another hard-to-treat brain tumor. They implanted human
brain metastatic and apoptosis-resistant VM-48 melanoma cells into the brains of immunodeficient
mice. Animals that were treated with narciclasine (1 mg/kg, orally) showed a significant
therapeutic benefit that was even slightly stronger than that of the established chemotherapeutic
agent temozolomide. The authors investigated the action of narciclasine on these melanoma
cells in detail and discovered a new direct target of the alkaloid, the eukaryotic
translation elongation factor eEF1A [49]. Binding of narciclasine to eEF1A was predicted by molecular docking analysis and
proven in a cell-free system with recombinant human eEF1A as well as in a cellular
assay using two melanoma cell lines. eEF1A is a very interesting protein: On the one
hand, it delivers aminoacyl-tRNAs to the empty A-site of ribosomes, on the other hand,
it binds to actin and participates in the organization of the actin cytoskeleton [50]. Thus, it regulates the morphology, cytokinesis, and migration of cells. Surprisingly,
although migratory processes in cancer cells are inhibited by narciclasine [48], a collagen invasion assay revealed that the invasive capacity of human cervix carcinoma
cells (HeLa) was not influenced by narciclasine at a concentration of 50 nM [51]. This might be due to the fact that cancer cell invasion does not only consist of
a migratory component. Another important factor is the interaction of the tumor cell
with the extracellular matrix.
Anti-inflammatory actions
The first report on its anti-inflammatory properties was published in 1999: Mikami
et al. [52] demonstrated that narciclasine effectively prevents paw swelling in a rat arthritis
model. They also showed that narciclasine suppresses the production of TNF-α in LPS-activated murine macrophages. The authors ascribed this effect to the protein
synthesis-blocking action of narciclasine [53]. In 2011, the alkaloid was reported to inhibit LPS-triggered NO production in a
murine macrophage cell line (RAW264) as well as the generation of TNF-α in a human monocytic cell line (THP-1) [54]. One year later, Lubahn et al. [55] provided evidence that narcistatin, a water-soluble cyclic phosphate prodrug of
narciclasine (see below), was able to reduce inflammation (by approx. 70 %) and joint
destruction (by approx. 50 %) in rat adjuvant-induced arthritis after disease onset.
Moreover, narcistatin also decreased the production of pro-inflammatory cytokines
in different types of leukocytes. In 2015, our group characterized the anti-inflammatory
effect of a narciclasine-containing extract of Haemanthus coccineus in two murine models of inflammation (dermal edema formation by arachidonic acid
or croton oil and kidney injury caused by unilateral ureteral obstruction) [56]. The extract blocked the pro-inflammatory activation of leukocytes and endothelial
cells. Interestingly, we could not detect any action on the activation cascade of
the most prominent pro-inflammatory transcription factor NFκB, but found a strong inhibition on the NFκB-dependent gene transcription in endothelial cell. We are currently performing further
experiments in order to fully understand the anti-inflammatory potential and underlying
mechanisms of the alkaloid.
Further pharmacological activities
In 1992, data on the antiviral actions of narciclasine were published. The compound
was found to be active in vitro against three flaviviruses (Japanese encephalitis, yellow fever, and dengue fever),
but not against the human immunodeficiency virus 1 (HIV-1) and the vaccinia virus.
However, the activity was very weak and the concentrations needed were too close to
those inducing cytotoxicity in uninfected cells [57].
More recently, in 2012, the influence of narciclasine on the circadian clock of cells
was investigated. The alkaloid reversibly extended the circadian period. This effect
was not caused by the inhibition of protein translation, but by an altered transcription
of the core clock gene Bmal1
[58].
Interestingly, in 2015, Kim et al. [59] investigated the action of narciclasine in the context of the Alzheimerʼs disease.
The alkaloid decreased the production of amyloid beta (Aβ) by attenuating amyloid precursor levels in vitro. In a murine Alzheimer model, a narciclasine-containing extract from the plant Lycoris chejuensis reduced the levels of Aβ and plaques, and showed beneficial effects on cognitive functions.
Narciclasine as a Lead Compound
Narciclasine as a Lead Compound
By analyzing the action of several derivatives, first insights into the structure-activity
relationship of narciclasine were already established in the 1970s. The tri-hydroxylated
ring C seems to be a critical part of the molecule ([Fig. 4]): Hydrophobic substituents decreased the inhibitory activity on peptide bond formation
and reduced the cytotoxicity against cancer cells [60], [61]. A loss of each of the three hydroxyl groups also attenuated the biological activity
[62], [63], [64]. Interestingly, after reduction of the double bond between C-1 and C-10b ([Fig. 4]), the resulting cis-dihydronarciclasine showed a weaker activity, whereas the trans-derivative was as active as narciclasine [51]. Pettit et al. [65] analyzed the biological activity of 7-deoxynarciclasine and 7-deoxy-trans-dihydronarciclasine ([Fig. 4]). Both modifications diminished the cytotoxic activity against tumor cells. A huge
number of narciclasine derivatives were synthesized by the group of R. Kiss [46]: Narciclasine was modified by the systematic addition of different substituents
to each of the hydroxyl groups (C-2/3/4), to the phenolic OH group (C-7) and to the
N atom of the lactam. In addition, the O atom of the lactam function was exchanged
with a C atom ([Fig. 4]). Unfortunately, most of these modifications led to a weaker antiproliferative activity.
Also the S-configuration at C-2 ([Fig. 4]) was shown to be of importance for the actions of the alkaloid [46]. Only narciclasine glucosylated at the phenolic group at C-7 ([Fig. 4]) exhibited an in vivo activity comparable to that of the parent compound, but showed a slightly increased
bioavailability. Taken together, the medicinal chemistry approach to optimize the
biological activity of narciclasine did not generate a largely improved compound.
 Fig. 4 Summary of the structural modifications of narciclasine and their impact on the biological
activity.
Fig. 4 Summary of the structural modifications of narciclasine and their impact on the biological
activity.
Nevertheless, an important physicochemical property of narciclasine, its poor water
solubility, could be improved: The group of G. Pettit converted narciclasine into
the water soluble (4 mg/mL) prodrug narcistatin ([Fig. 4]), a cyclic phosphate bridging the two hydroxyl groups at C-3 and C-4. Narcistatin,
which is readily hydrolyzed to narciclasine by unspecific esterases, showed the same
pharmacological potency as the parent compound [66].
The group of R. Kiss also evaluated both the pharmacokinetic properties and the adverse
effects of narciclasine. In mice, the oral administration of 10 mg/kg resulted in
a peak concentration of 300 ng/mL. The oral bioavailability was calculated as 32 %,
the terminal elimination half-life after iv application was 66 min [46]. Of note, the alkaloid was suggested to be able to cross the blood brain barrier,
since brain tumors in mice were successfully treated [48], [49]. Using liver microsomes from different species (rat, mouse, dog, human), narciclasine
was found to be metabolically quite stable: After 45 min, only the rodent species
evoked a slight decline in the concentration of narciclasine of less than 20 % [46]. However, the observed actions in the brain are not necessarily caused by the compound
itself. Its physicochemical properties are not favorable for the permeation of the
blood brain barrier. Thus, unknown metabolites of narciclasine could be responsible
for these actions. Unfortunately, this interesting issue has not been investigated
so far. McNulty et al. [67] reported an inhibitory activity of the alkaloid against the human cytochrome P450
isoenzyme CYP3A4, which points towards an unfavorable interaction profile with a great
number of drugs that serve as a substrates of this enzyme.
The side effects of narciclasine were studied in rats: The compound was administered
by oral gavage for five consecutive days (Monday to Friday) for 3 weeks at doses of
0, 1, 3, 10, and 25 mg/kg/day. The no observed adverse effect level (NOAEL) was 1 mg/kg/day.
Higher doses (3 and 10 mg/kg/day) caused piloerection, diarrhea, lethargy, stomach
abnormalities and alterations in red and white blood cell parameters. The highest
dose was too toxic and caused death in most of the animals within three days [46].
Summary
Taken together, narciclasine shows strong cytotoxic activity (nanomolar range) against
a variety of tumor cells in vitro. The in vivo efficacy was very good in brain tumor models, whereas the action was not very pronounced
in other types of cancer. Two direct targets of narciclasine have been discovered
so far: ribosomes and eEF1A. Recent findings suggest that the actin cytoskeleton and
thus processes like cytokinesis and cell migration are disturbed by narciclasine.
This new knowledge has challenged the long standing concept of narciclasine primarily
acting as an inhibitor of protein biosynthesis. Beyond cancer, a second promising
field are inflammatory diseases, since the alkaloid has emerged to exhibit profound
antiphlogistic properties in vitro and in vivo. Unfortunately, in particular in the area of inflammation, but also in the much better
investigated field of cancer, only few studies provided in-depth insights into altered
cellular functions and the underlying molecular mechanism. Moreover, the development
of narciclasine is hampered by a lack of efficient supply, since total syntheses are
complex and biotechnological approaches are completely missing. Thus, despite all
the intriguing findings about narciclasine, it still remains inconclusive whether
narciclasine could be advanced to clinical trials.





