Definition, Inzidenz, klinisches Bild
Definition, Inzidenz, klinisches Bild
Das Plasmazellmyelom (WHO-Definition) wird in der klinischen Routine meist als Multiples
Myelom (MM) bezeichnet. Das MM ist eine maligne Erkrankung der Plasmazellen, welche
die am weitesten differenzierten Zellen der B-Lymphozyten-Reihe sind. Durch die monoklonale
Plasmazellvermehrung im Knochenmark werden komplette und/oder inkomplette Immunglobuline
(Leichtketten) vermehrt produziert. Die monoklonalen Immunglobuline lassen sich im
Serum und/oder Urin als „M-Gradient“ ([Abb. 1]) bzw. im Serum mittels des freien Leichtkettentests quantifizieren. Das symptomatische
MM ist eine Systemerkrankung und durch multiple Osteolysen, Nierenfunktionsverschlechterung,
Anämie und/oder Hyperkalzämie gekennzeichnet. Seit 2014 werden auch Biomarker für
die Diagnosesicherung des symptomatischen MM genutzt ([Tab. 1]). Abzugrenzen ist das solitäre Plasmozytom, welches durch einen isolierten ossären
oder extraossären Plasmazelltumor definiert ist.
 Abb. 1 Beispiele für M-Gradienten in der Elektrophorese bei a monoklonaler Gammopathie unklarer Signifikanz, b Smoldering Multiplem Myelom und c multiplem Myelom.
Abb. 1 Beispiele für M-Gradienten in der Elektrophorese bei a monoklonaler Gammopathie unklarer Signifikanz, b Smoldering Multiplem Myelom und c multiplem Myelom.
Tab. 1
Diagnosekriterien für die Monoklonale Gammopathie unklarer Signifikanz (MGUS), das
Smoldering Multiple Myelom und das Multiple Myelom der International Myeloma Working
Group [2]
[3].
|
MGUS
|
Smoldering MM
|
MM (behandlungspflichtig)
|
|
monoklonales Protein
|
< 30 g/l im Serum
|
≥ 30 g/l im Serum,
|
vorhanden im Serum und/oder Urin
|
|
und
|
und/oder
|
|
|
< 500 mg/Tag im Urin
|
> 500 mg/Tag im Urin
|
|
|
und
|
und/oder
|
und/oder
|
|
prozentualer Anteil der monoklonalen Plasmazellen im Knochenmark
|
< 10 %
|
≥ 10 % – 60 %
|
≥ 10 % oder Plasmozytom
|
|
und
|
und
|
und
|
|
Organschädigung
|
keine
|
keine
|
SLIM-CRAB-Kriterien erfüllt
|
SLIM-CRAB-Kriterien
Endorganschäden (mindestens eines der folgenden vier): C = Hyperkalzämie (Konzentration
im Serum > 11 mg/dl oder 0,25 mmol/l über dem Normwert); R = Niereninsuffizienz (Kreatinin
> 2 mg/dl oder Kreatinin Clearance < 40 ml/min); A = Anämie (Hämoglobin < 10 g/dl
oder > 2 g/dl unter dem Normwert); B = Knochenerkrankung (eine oder mehrere Osteolysen
nachgewiesen durch Projektionsradiografie, CT oder PET-CT).
und/oder
Biomarker (mindestens ein Biomarker nachweisbar): Anteil der klonalen Plasmazellen
im Knochenmark ≥ 60 %, Verhältnis von beteiligten zu unbeteiligten freien Leichtketten
im Serum ≥ 100 (Werte basieren auf dem serum Freelite assay von Binding Site) und
betroffene freie Leichtketten mit einer Konzentration von ≥ 100 mg/l, mehr als eine
fokale Läsion im MRT ≥ 5 mm.
Das MM ist die zweithäufigste hämatologische maligne Erkrankung in den westlichen
Industriestaaten. Die Inzidenz liegt bei ca. 6 Neuerkrankungen pro 100 000 Einwohner
in Europa [1]. Patienten sind zum Zeitpunkt der Diagnose im Median ca. 70 Jahre alt. Die mittlere
Überlebenszeit konnte in den letzten 15 Jahren verdoppelt werden. So überleben Patienten
nach Hochdosistherapie im Median 8 – 10 Jahre; Patienten, die nicht intensiv behandelt
werden können ca. 5 Jahre.
Die Symptome des MM sind unspezifisch. 80 % der Patienten klagen über Knochenschmerzen.
Weitere häufige Symptome sind Anämie (50 %), Nierenfunktionsverschlechterung (20 %)
und Hyperkalzämie (bis zu 15 %). Die Zeit bis zur Diagnosesicherung nach dem Auftreten
der ersten Symptome beträgt im Median 3 Monate. Alle Patienten mit MM haben in Ihrer
Vorgeschichte eine Monoklonale Gammopathie unbestimmter Signifikanz (MGUS) und/oder
ein Smoldering Multiples Myelom (SMM), welche aber aufgrund fehlender Symptomatik
oftmals nicht diagnostiziert wurden. Die Differenzierung zwischen MGUS, SMM und MM
ist in [Tab. 1] dargestellt.
Bei unspezifischen Symptomen wie Knochenschmerzen, Anämie, Nierenfunktionsverschlechterung
und/oder Hyperkalziämie sollte ein MM ausgeschlossen werden.
Diagnostik
Für die Diagnosesicherung des MM sind serologische Untersuchungen, eine Knochenmarkpunktion
bzw. eine Tumorbiopsie und eine Skelettbildgebung erforderlich (Box 1). Die Kriterien
der International Myeloma Working Group zur Diagnose des MM wurden 2014 aktualisiert
[2]
[3]. Bei dieser Aktualisierung wurden die klassischen CRAB-Kriterien um sogenannte „Biomarker“
zu den heute gültigen SLIM-CRAB-Kriterien erweitert ([Tab. 1]). Diese Biomarker kündigen mit einer Wahrscheinlichkeit von > 80 % das Auftreten
von Symptomen, bedingt durch Organschäden, innerhalb der folgenden 2 Jahre an. Die
Diskussion hinsichtlich Überbehandlung versus Kuration des MM im frühen Stadium wird
aktuell kontrovers geführt. Deshalb sollten Patienten mit SLIM-CRAB-Kriterien möglichst
in prospektiven Registern oder Therapiestudien erfasst bzw. behandelt werden. Das
SMM ist nach den aktuellen Kriterien nicht zu behandeln. Weltweit sind Studien aktiv,
welche den sehr frühen Therapiebeginn beim SMM prospektiv untersuchen.
Labor- und Knochenmarkdiagnostik sowie Bildgebung beim Multiplen Myelom zum Zeitpunkt
der Diagnosestellung
Labordiagnostik
-
peripheres Blut: Blutbild mit Differenzialblutbild
-
biochemische Parameter: Elektrolyte (Natrium, Kalium, Kalzium), glomeruläre Filtrationsrate,
Kreatinin, Harnstoff, Harnsäure, C-reaktives Protein (CRP), Laktatdehydrogenase (LDH)
-
Beta-2-Mikroglobulin, Gesamteiweiß, Albumin, Immunglobuline quantitativ (IgA, IgG,
IgM), Glutamat-Oxalacetat-Transaminase (GOT), Glutamat-Pyruvat-Transaminase (GPT),
Bilirubin, alkalische Phosphatase (AP)
-
Serumproteinelektrophorese, Immunfixationselektrophorese
-
Bestimmung freier Leichtketten im Serum (Freelite® Test)
-
Urinuntersuchungen (24 Stunden Sammelurin): Kreatinin-Clearence, Urinproteinelektrophorese
oder quantitative Leichtkettenbestimmung (kappa und lambda), Immunfixationselektrophorese,
Gesamtprotein, Albumin
Knochenmarkdiagnostik
-
Aspirationszytologie
-
Histologie (mit immunhistochemischer Sicherung der Leichtkettenrestriktion)
-
molokulare Zytogenetik an CD138-angereicherten Plasmazellen mithilfe der Fluoreszenz
In-situ-Hybridisierung (FISH) z. B.: t(4;14), t(14;16), del(17p13), + 1q21 > 3 Kopien
→ ungünstige Prognose
Bildgebung
-
Low-dose-Ganzkörper-Computertomographie (ohne Kontrastmittel) zur Diagnostik von Osteolysen,
Osteopenie und zur Stabilitätsbeurteilung
-
MRT bei Verdacht auf extramedulläre Manifestationen und zur Abklärung der Behandlungsbedürftigkeit
eines SMM
Hinsichtlich der Bildgebung ist zu betonen, dass in Deutschland die Niedrig-Dosis-CT
weitgehend flächendeckend realisierbar ist. Die MRT kann bei frühen Stadien und negativem
Niedrig-Dosis-CT-Befund weiterführende Informationen erbringen. Insbesondere fokale
Läsionen im Knochenmark korrelieren mit einer schnellen Symptomentwicklung. Deshalb
sind eindeutige multiple fokale Läsionen in der MRT eine Behandlungsindikation.
Für die Knochenmarkdiagnostik ist eine Klassifikation der Zellen in unreife oder reife
Plasmazellen zu empfehlen. Die aus dem Knochenmark aspirierten Myelomzellen sind insbesondere
bei Eignung für eine intensive Therapie hinsichtlich zytogenetischer Aberrationen
zu untersuchen. Prognostisch ungünstige zytogenetische Aberrationen sind 4;14- oder
14;16-Translokationen, die 17p-Deletion oder das Vorliegen von mehr als 3 Kopien der
1q21-Amplifikation. Erstmalig werden Therapien an zytogenetische Risikomuster gekoppelt.
Zum Beispiel profitieren Hochrisiko-Patienten von einer langandauernden Proteasominhibitor-basierten
Therapie [4].
Um die Prognose zu beschreiben, sind verschiedene Stadieneinteilungen des MM publiziert.
Die am längsten etablierte Einteilung ist die Stadieneinteilung nach dem International
Staging System (ISS) basierend auf den Serumwerten für Albumin und Beta-2-Mikroglobulin.
Zu diesen serologischen Parametern kann die Zytogenetik addiert werden, sodass eine
bessere Diskriminierung der Risikogruppen erfolgt. Eine weitere Möglichkeit ist die
zusätzliche Implementierung des LDH-Werts, sodass mit dem revidierten R-ISS eine zuverlässige
Prognoseabschätzung möglich ist [5].
Erstlinientherapie
Symptomatische Myelompatienten sind umgehend zu behandeln. Patienten mit positiven
Biomarkern als alleinigem Therapiekriterium sind über die Prognose und mögliche Komplikationen
aufzuklären. In der klinischen Praxis kann bei Unsicherheit über die Therapieindikation
abgewartet werden. So sind bei unklaren Läsionen in der Kernspintomografie Folgeuntersuchungen
nach 3 – 6 Monaten sinnvoll. Treten neue Läsionen auf oder vergrößern sich bekannte
Läsionen, so ist die Therapieindikation als gesichert anzusehen. Bei Patienten mit
einem Leichtkettenquotienten ≥ 100 und einer absoluten Konzentration der beteiligten
Leichtkette von ≥ 100 mg/dl sollte eine erneute Messung im Verlauf zum Ausschluss
von Messungenauigkeiten erfolgen. Patienten mit hoher Compliance (d. h. Patienten,
die mehr als 2 Liter am Tag trinken, keine nicht-steroidalen Antirheumatika einnehmen
und bei Fieber frühzeitig den Arzt aufsuchen) können beobachtet werden. So sind Patienten
bekannt, die aufgrund eines positiven SLIM-Kriteriums als behandlungspflichtig anzusehen
sind, bei denen über viele Jahre kein Endorganschaden aufgetreten ist.
Bei einer positiven Therapieentscheidung ist umgehend mit dem Patienten zu besprechen,
welche Therapieoption gewählt wird. Zu unterscheiden ist zwischen der intensiven Behandlung,
welche eine Hochdosistherapie gefolgt von einer autologen Blutstammzelltransplantation
beinhaltet und einer Behandlung ohne Hochdosistherapie. Ein transplantationserfahrener
Kollege sollte möglichst in die Entscheidung einbezogen werden, da durch den Patienten
die Risiken der Hochdosistherapie oft als sehr belastend angesehen werden. Einen Überblick
über die primäre Therapieentscheidung gibt [Abb. 2].
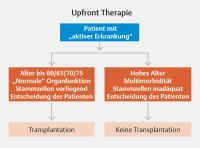 Abb. 2 Entscheidungskriterien zur Wahl der Erstlinientherapie.
Abb. 2 Entscheidungskriterien zur Wahl der Erstlinientherapie.
Für fitte Patienten bis zum Alter von 70 Jahren, wird heute die Hochdosistherapie
mit 200 mg/m² Melphalan empfohlen. Die Tumormassenreduktion vor Hochdosistherapie
erfolgt durch eine Induktionstherapie. Hier werden Dreierkombinationen bevorzugt.
In Deutschland ist eine Kombination mit Bortezomib/Cyclophosphamid/Dexamethason oder
Bortezomib/Lenalidomid/Dexamethason (im Rahmen der integrierten Versorgung umsetzbar)
als beste Option anzusehen. Nach der Stammzellsammlung wird einmalig transplantiert.
Gegenwärtig ist die Diskussion über die Tandemtransplantation nicht abgeschlossen.
Wir empfehlen bei Nichterreichen mindestens einer nahezu kompletten Remission außerhalb
von Studien eine 2. Hochdosistherapie, um die Remissionstiefe zu erhöhen.
Die Therapieoptionen nach Hochdosistherapie werden unterschiedlich gewertet. Außerhalb
von Studien sollte für Patienten mit Resterkrankung und günstiger Zytogenetik eine
Erhaltungstherapie erfolgen. Die Behandlung mit Thalidomid 50 mg wird in diesem Fall
von den Krankenkassen in Deutschland erstattet. Patienten mit zytogenetischer Hochrisikokonstellation
sollten über zwei Jahre mit Bortezomib 1,3 mg/m² alle zwei Wochen behandelt werden,
hier muss die Kostenübernahme jedoch bei der Krankenkasse beantragt werden.
Für Patienten, die älter als 70 Jahre sind, wird in der Regel mit modernen Medikamenten
primär behandelt. Für Patienten in gutem Allgemeinzustand ist eine Dreierkombination,
z. B. mit Bortezomib/Melphalan/Prednisolon oder Bortezomib/Cyclophospamid/Dexamethason
eine gute Option. Für Patienten in eingeschränktem Allgemeinzustand ist die weniger
belastende Langzeittherapie mit Lenalidomid/Dexamethason als Primärtherapie zu empfehlen.
Teile der Erstlinientherapie beim MM sollten an Risiko- und Remissionsstatus des Patienten
angepasst werden.
Rezidivtherapie
Meist wird ein Krankheitsprogress oder ein Rezidiv laborchemisch diagnostiziert. Bei
einer Verdopplung des monoklonalen Proteins in kurzer Zeit (ca. 3 Monate) sollte mit
einer systemischen Therapie begonnen werden. Werden CRAB-Kriterien nachgewiesen, so
ist die Rezidivtherapieindikation eindeutig gegeben. Die Entscheidung, wann mit der
Rezidivtherapie bei serologisch progredienten Patienten begonnen werden sollte, ist
individuell zu treffen. Organschädigungen (z. B. Frakturen) sollten vermieden werden.
Die Wahl der Rezidivtherapie erfolgt hauptsächlich anhand von Patientenfaktoren, wie
Alter, Komorbiditäten und Patientenpräferenzen sowie unter Berücksichtigung der Vortherapien
(Effektivität, Nebenwirkungen, Dauer des Therapieansprechens). In den letzten 2 Jahren
wurden erfreulicherweise 5 neue Substanzen zur Rezidivtherapie des MM zugelassen ([Tab. 2]) [6]. Eine gesicherte Sequenz der Rezidivtherapien kann im Augenblick nicht empfohlen
werden, da kein „Head-to-head-Vergleich“ der neuen Substanzen umfassend erfolgt ist.
Eine Ausnahme stellt Carfilzomib dar, welches eine therapeutische Überlegenheit gegenüber
Bortezomib in der Endeavor-Studie zur Rezidivtherapie zeigte.
Tab. 2
Beispiele für neu zugelassene Arzneimittel oder Erweiterung der Zulassung (Indikation
MM) seit 2015.
|
Wirkstoff
|
Indikation
|
|
Carfilzomib
|
Kombination mit Dexamethason/Lenalidomid oder Dexamethason mono zur Behandlung von
erwachsenen Patienten mit MM, die mindestens eine Vortherapie erhalten haben.
|
|
Lenalidomid
|
Patienten mit unbehandeltem MM, die nicht transplantierbar sind. Kombination mit Dexamethason
bei Patienten, die mindestens eine vorausgegangene Therapie erhalten haben.
|
|
Panobinostat
|
Kombination mit Dexamethason und Bortezomib bei Patienten, mit mindestens zwei Vortherapien,
darunter Bortezomib und eine immunmodulierende Substanz.
|
|
Daratumumab
|
Monotherapie bei Erwachsenen mit rezidiviertem und refraktärem MM, die bereits mit
einem Proteasominhibitor und einem Immunmodulator behandelt wurden und welche während
der letzten Therapie Krankheitsprogression zeigten.
|
|
Elotuzumab
|
Kombination mit Dexamethason und Lenalidomid bei erwachsenen Patienten, die mindestens
eine Vortherapie erhalten haben.
|
|
Ixazomib
|
Kombination mit Lenalidomid und Dexamethason für die Behandlung von erwachsenen Patienten,
die mindestens eine vorherige Therapie erhalten haben.
|
Die Auswahl der Rezidivtherapie sollte in Abwesenheit evidenzbasierter Sequenzen unter
Berücksichtigung von Patientenfaktoren und Vortherapien erfolgen.
Ausblick
Die klonale Genese des MM wird intensiv untersucht. Aufbauend auf dem erweiterten
Verständnis der Pathogenese werden neue Therapieformen entwickelt. Auch beim MM wird
die Remissionstiefe durch neue Medikamente oder Medikamentenkombinationen gesteigert.
Begleitend sind Methoden der Messung der sogenannten minimalen Resterkrankung (MRD,
minimal residual disease) weiterzuentwickeln. Die klassische Chemotherapie zur Behandlung
des MM wird weitgehend verlassen. Neue Therapieoptionen wie Proteasominhibiton, Deacetylase-Hemmung
und Immunmodulation haben zunehmend an Bedeutung gewonnen. Zudem hat die Verfügbarkeit
von Antikörpern zur Therapie des MM das therapeutische Arsenal entscheidend erweitert.
Weitere Formen der Immuntherapie sind in frühen Studien mit erfolgversprechenden Resultaten
gestartet. Es ist zu erwarten, dass die „Immun-Onkologie“ auch beim MM einen wichtigen
Schritt in Richtung Heilung beitragen wird.
Die bereits zugelassenen neuen Therapeutika haben zu einer eindrücklichen Prognoseverbesserung
für Patienten mit MM geführt; die konsequente Weiterentwicklung neuer Therapieansätze
stimmt für die Zukunft weiter optimistisch.





