
Subscribe to RSS
DOI: 10.1055/s-0038-1677461
Commentary: Controversies in Thrombosis and Hemostasis Part 2–Does Sticky Platelet Syndrome Exist?
Publication History
Publication Date:
10 January 2019 (online)

Hemostasis = love.
Everyone talks about, but no one understands it.
Hemostasis is complex. The process of hemostasis has several outcomes, including physiologically arresting blood loss at sites of vascular injury. In simple terms, this is achieved by formation of a “plug that seals the hole.” This “plug” comprises a framework of blood components that act together to create a stable mass that stops further blood leakage.
Hemostasis involves the complex interaction of many integrated processes.[1] Central to hemostasis is the blood milieu, containing platelets, which in part act like “bricks,” and many adhesive plasma proteins, which in part act like “mortar,” working together to develop a framework that we sometimes call the “platelet plug.” However, neither the platelets nor the plasma proteins are passive agents in this process. When hemostasis works, everything is in physiological balance, and we are not even cognizant of this activity taking place in our own bodies. When hemostasis gets out of balance, however, disease or pathological processes can occur.
Plasma is complex, but platelets are even more complicated.[2] [3] This journal has published several papers on platelet physiology, and the ongoing popularity of these papers attests to the complexity of platelet physiology and their interest in our readership.[4] [5] Each individual platelet expresses a huge number of proteins on their surface that we call platelet receptors, and these interact with many plasma components including other proteins. There are around 100 million platelets in each mL of our blood.
Hemostasis can be conveniently separated into primary and secondary hemostasis, with primary hemostasis reflecting predominantly vessel wall and platelet activity (including adhesion), and secondary hemostasis mainly reflecting the plasma coagulation system (including clot formation).[1] Although this provides a convenient separation to help teach hemostasis to junior scientists, and to study this process in vitro, it needs to be recognized that, in vivo at least, these systems do not work in isolation. Primary and secondary hemostasis work in concert to form the platelet plug. Injury to a blood vessel causes a cascade of events. The injury releases cellular components including “tissue factor,” as well as exposing subendothelial surfaces that are normally not exposed to this blood. This causes activation of “secondary hemostasis” by several mechanisms. Sometimes we use terms such as the “coagulation cascade,” but the important elements here are generation of thrombin and conversion of soluble plasma protein fibrinogen to “insoluble” fibrin. The vessel injury also leads to platelet activation and platelet adhesion to the damaged site. Integral to the process of platelet adhesion are the platelet receptors and many plasma proteins, including von Willebrand factor (VWF), which act as a kind of sticky string to “glue” the platelets together and fasten them to the damaged subendothelial surface.[6] Platelets aggregate to form the scaffolding of the platelet plug, and the entire structure is further stabilized by the fibrin being formed, as robustly integrated into the platelet plug.
Indeed, much hemostasis activity goes into creating the platelet plug. In normal physiology, this platelet plug seals the hole, and thus permits wound healing to subsequently occur. However, if the process of hemostasis is dysfunctional, pathological situations can ensue. On one side of this unbalanced hemostasis is where hemostasis may be “under-active” and patients can bleed (e.g., if the plasma components or platelets do not work as well as they should, the formation of the platelet plug may be delayed, or else it may not be formed properly). If hemostasis is “over-active,” patients may be at increased risk of thrombosis. In such situations, the platelet plug may grow too large and block off a blood vessel. If this occurs in a venous vessel in the leg, it may cause a deep vein thrombosis (DVT), which (should some of the thrombosis break off and travel to the lung to block off some of its blood vessels) can then lead to the development of pulmonary embolism. If the thrombosis-related blockage occurs in the heart, it may lead to a heart attack, and if it occurs in the brain, it may cause an ischemic cerebral infarction (i.e., stroke). If the thrombosis occurs in a placenta, it can cause pregnancy morbidity.
There is a general belief that platelets, along with the plasma protein VWF, represent more important hemostasis components in the arterial blood system, due to the importance of shear stress in VWF/platelet function. This concept is supported by evidence that arterial thrombi are more “enriched” in platelets, because the risk of arterial vascular events can theoretically be prevented by use of antiplatelet agents such as aspirin.[7] Conversely, there is a general belief that the plasma coagulation components (i.e., secondary hemostasis) are more important in the venous blood system. This concept is supported by evidence that venous thrombi are more “enriched” in fibrin, because the risk of venous vascular events can theoretically be prevented by use of anticoagulation agents such as heparins, vitamin K antagonist drugs (e.g., warfarin), or direct oral anticoagulants. Additional evidence includes that coagulation factor (F) deficiencies (e.g., FVIII or FIX, leading to hemophilia) primarily cause bleeding in venous vessels (e.g., hematomas), and that deficiencies in natural anticoagulants (e.g., protein S, protein S, antithrombin) more frequently cause thrombosis in venous vessels (e.g., DVT).
However, these concepts oversimplify hemostasis. Although primary and secondary hemostasis can be evaluated separately in vitro, simply by separating out the components, the process is intertwined in vivo, with clear overlaps or “shades of gray.” For example, more detailed investigation of venous thrombi will uncover the presence of platelets, and more detailed investigation of arterial thrombi will uncover the presence of fibrin.[7] Similarly, sometimes antiplatelet agents are prescribed to prevent thrombotic complications that occur outside the arterial bed.
Workers in hemostasis in general accept the concept that deficiencies of coagulation proteins can lead to bleeding, that deficiencies of natural anticoagulants can cause venous thrombosis, and that venous thrombosis can be prevented by anticoagulant therapy. Workers in hemostasis also accept the concept that deficiencies of platelets, or that the existence of platelet dysfunction, can lead to bleeding. Workers in hemostasis even accept that these events may have a congenital or acquired basis.
Workers in hemostasis have also started to accept additional concepts, for example, that excesses in some coagulation proteins can increase the risk of thrombosis—this is now accepted for FVIII, and starting to be accepted for VWF. However, workers in hemostasis seem to have mixed feelings about the involvement of overactive platelets in thrombosis and any arising pathophysiology. On the one hand, most will accept that treatment of patients at risk of arterial thrombosis with antiplatelet agent will help to lower such risk. On the other hand, some workers in the field seem to be less prone to accept the presence of “hyperactive” platelets, or at least that congenital forms of “hyperactive” platelets may lead to thrombosis.
This journal has published several recent articles on the condition popularly called “sticky platelet syndrome (SPS).”[8] [9] [10] This is believed, by those who deem the condition to exist, to represent platelet hyperactivity having a familial tendency (i.e., congenital or inherited) in at least some cases. The commonly proposed pathophysiological outcome includes thrombosis, namely venous thrombosis events, and pregnancy morbidity. Indeed, the current issue of this journal includes one such paper, another update review on SPS.[11] We hope that users of Seminars in Thrombosis and Hemostasis (STH) read this paper with an open mind.
In this commentary, we ask the question “Does SPS exist?,” because we know that many of our hemostasis colleagues have doubts about its existence. We know they have such doubts because they tell us so, and sometimes reflect surprise that STH seems to publish periodically on SPS, when other major hemostasis journals do not, and when many in the hemostasis field do not even believe SPS to exist.
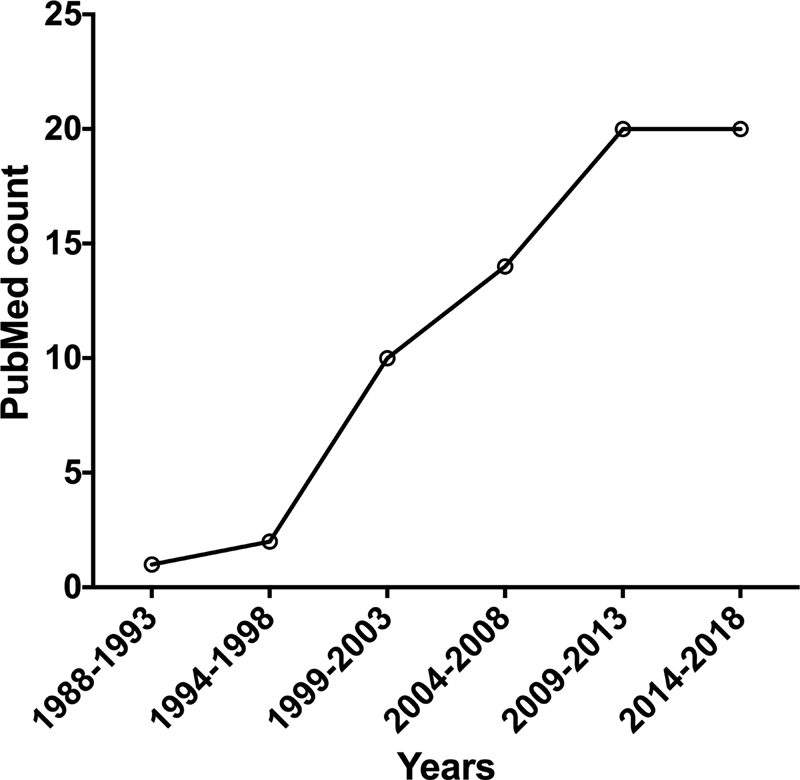
Just as primary and secondary hemostasis is intertwined, so too is the history of SPS and STH. Indeed, the founding Editor-in-Chief of STH, Eberhard F. Mammen, was a strong advocate for SPS, and his early works (including the first article ever published using the moniker of “sticky platelet syndrome” according to a PubMed search of this term)[12] [13] have formed the foundation of current research in the field.[9] [10] [11] Does STH really publish a lot of papers on SPS? A recent PubMed search of the term “sticky platelet syndrome” identified a total of 63 papers, 8 of which were published in STH. Another journal publishing in the area of thrombosis and hemostasis has published more of these papers, at a count of 13. But there were no papers in the highest impact factor hematology journals.
In any case, SPS seems to be building up some sort of belief base, if we accept that the increasing publication rate identified by our PubMed search ([Fig. 1]) represents increasing acceptance of SPS as a prothrombotic tendency among peers.
Why is SPS plausible, and why is SPS alternatively plausibly deniable? Unless a worker in the field of research has actively investigated a condition, that worker may have a healthy skepticism about whether a particular condition exists. We explored this in an earlier commentary on hematidrosis.[14] Accordingly, unless a worker in the field of thrombosis research has actively investigated SPS as a prothrombotic condition, that worker may have a healthy skepticism about whether SPS really exists as an autonomous clinical entity. Investigating SPS, and indeed platelet-related research, is problematic. Unlike plasma-based hemostasis research work, the study of SPS and platelets requires fresh patient material and highly meticulous sample collection and handling. In other words, the patient under investigation requires blood collection at the time of investigation, and then platelet function testing needs to be performed immediately afterwards by experienced personnel.
Neither of the authors to this commentary currently undertake extensive platelet research. This is not because we are not capable of performing this research, but platelet research is very challenging and we perhaps undertook “other/easier/more-productive/less debated” paths. Accordingly, neither of us has ever systematically investigated SPS as an entity within our routine laboratory activity. Indeed, most workers in hemostasis research have never systematically investigated SPS. Nonetheless, we tend to believe that hyperactive platelets can cause prothrombotic events and induce pregnancy morbidity. We also believe that there is at least some element of genetics or inheritability in such platelet hyperactivity, at least on occasion. So, call it hyperactive platelets or “sticky platelet syndrome,” we do not doubt the plausibility of hyperactive primary hemostasis leading to adverse pathophysiology.[15] There are many examples of “acquired” conditions leading to activated platelets and subsequent thrombosis—including heparin-induced thrombocytopenia.[16] [17] Of course, SPS may not be a single discrete condition—just as hemophilia is not just a single discrete pathological bleeding condition reflective of secondary hemostasis.
We are also reminded that there are several precedents for inherited bleeding conditions caused by genetic changes in platelet receptors, for example, the glycoprotein Ib receptor, which acts as the primary VWF binding receptor on platelets. That condition is called “platelet-type von Willebrand disease” (PT-VWD), and in essence creates “hyperactive” platelets that lead to loss of high molecular weight forms of VWF, and usually also (mild) thrombocytopenia.[18] No one in the hemostasis field doubts in the existence of PT-VWD, although perhaps counter-intuitively, PT-VWD presents as a bleeding disorder. PT-VWD is also a “cousin” (or nonidentical “twin”) to type 2B VWD, which represents a hyperactive form of VWF.[19] [20] [21] [22] Also, no one doubts the genetic basis of either PT-VWD or 2B VWD.
The update review in the current issue of this journal[11] provides other insights into why some workers in hemostasis may not believe in SPS. The purpose of this particular commentary is to encourage further dialogue, not only in regard to SPS but also as related to other potential controversies in thrombosis and/or hemostasis, as the situation may present in the future. It is acceptable to have healthy scientism. It is also acceptable to have an open mind.
Disclaimer
The opinions expressed in this commentary are those of the authors, and are not necessarily those of the University Hospital of Verona or of New South Wales Health Pathology.
-
References
- 1 Bonar RA, Lippi G, Favaloro EJ. Overview of hemostasis and thrombosis and contribution of laboratory testing to diagnosis and management of hemostasis and thrombosis disorders. Methods Mol Biol 2017; 1646: 3-27
- 2 Gremmel T, Frelinger III AL, Michelson AD. Platelet physiology. Semin Thromb Hemost 2016; 42 (03) 191-204
- 3 Preston RJS, Lisman T. Extrahemostatic functions of platelets and coagulation factors. Semin Thromb Hemost 2018; 44 (02) 89-90
- 4 Favaloro EJ. 2017 Eberhard F. Mammen Award Announcements: Part I-most popular articles. Semin Thromb Hemost 2017; 43 (04) 357-363
- 5 Favaloro EJ. 2018 Eberhard F. Mammen Award Announcements: Part I-most popular articles. Semin Thromb Hemost 2018; 44 (03) 185-192
- 6 Dorgalaleh A, Tabibian S, Shiravand Y, Favaloro EJ. von Willebrand disease. In: Dorgalaleh A. , ed. Congenital Bleeding Disorders – Diagnosis and Management. Cham, Switzerland: Springer Press; 2018
- 7 Lippi G, Favaloro EJ. Venous and arterial thromboses: two sides of the same coin?. Semin Thromb Hemost 2018; 44 (03) 239-248
- 8 Sokol J, Skerenova M, Jedinakova Z. , et al. Progress in the understanding of sticky platelet syndrome. Semin Thromb Hemost 2017; 43 (01) 8-13
- 9 Kubisz P, Ruiz-Argüelles GJ, Stasko J, Holly P, Ruiz-Delgado GJ. Sticky platelet syndrome: history and future perspectives. Semin Thromb Hemost 2014; 40 (05) 526-534
- 10 Kubisz P, Stasko J, Holly P. Sticky platelet syndrome. Semin Thromb Hemost 2013; 39 (06) 674-683
- 11 Kubisz P, Holly P, Stasko J. Sticky platelet syndrome – 35 years of growing evidence. Semin Thromb Hemost 2019; 45 (01) 61-68
- 12 Mammen EF, Barnhart MI, Selik NR, Gilroy J, Klepach GL. “Sticky platelet syndrome”: a congenital platelet abnormality predisposing to thrombosis?. Folia Haematol Int Mag Klin Morphol Blutforsch 1988; 115 (03) 361-365
- 13 Mammen EF. Sticky platelet syndrome. Semin Thromb Hemost 1999; 25 (04) 361-365
- 14 Favaloro EJ, Lippi G. Commentary: controversies in thrombosis and hemostasis part 1-hematidrosis: “Blood, Sweat and Fears” or A “Pigment of Fertile Imaginations?”. Semin Thromb Hemost 2018; 44 (03) 296-297
- 15 Salvagno GL, Pavan C, Lippi G. Rare thrombophilic conditions. Ann Transl Med 2018; 6 (17) 342
- 16 Favaloro EJ, McCaughan G, Pasalic L. Clinical and laboratory diagnosis of heparin induced thrombocytopenia: an update. Pathology 2017; 49 (04) 346-355
- 17 Favaloro EJ, McCaughan G, Mohammed S. , et al. HIT or miss? A comprehensive contemporary investigation of laboratory tests for heparin induced thrombocytopenia. Pathology 2018; 50 (04) 426-436
- 18 Othman M, Emsley J. Platelet-type von Willebrand disease: toward an improved understanding of the “sticky situation”. Semin Thromb Hemost 2014; 40 (02) 146-150
- 19 Othman M. Platelet-type von Willebrand disease and type 2B von Willebrand disease: a story of nonidentical twins when two different genetic abnormalities evolve into similar phenotypes. Semin Thromb Hemost 2007; 33 (08) 780-786
- 20 Favaloro EJ. Phenotypic identification of platelet-type von Willebrand disease and its discrimination from type 2B von Willebrand disease: a question of 2B or not 2B? A story of nonidentical twins? Or two sides of a multidenominational or multifaceted primary-hemostasis coin?. Semin Thromb Hemost 2008; 34 (01) 113-127
- 21 Othman M. Platelet-type von Willebrand disease: a rare, often misdiagnosed and underdiagnosed bleeding disorder. Semin Thromb Hemost 2011; 37 (05) 464-469
- 22 Othman M, Kaur H, Emsley J. Platelet-type von Willebrand disease: new insights into the molecular pathophysiology of a unique platelet defect. Semin Thromb Hemost 2013; 39 (06) 663-673