Subscribe to RSS
DOI: 10.1055/a-1331-2469
Seltene Erkrankungen der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis
Article in several languages: deutsch | EnglishAuthors

Zusammenfassung
Seltene Erkrankungen der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis stellen aufgrund der niedrigen Inzidenz und den daraus resultierend eingeschränkten diagnostischen Kriterien sowie therapeutischen Optionen eine Herausforderung dar. Der Schwellenwert, ab dem eine Erkrankung als selten zu werten ist, liegt bei höchstens 5 Betroffenen pro 10 000 Personen. Innerhalb dieser Erkrankungen gibt es allerdings extreme Schwankungen. So gehören einige seltene Erkrankungen, wie z.B. das invertierte Papillom, zu den regelmäßig diagnostizierten und therapierten Krankheitsbildern einer größeren Klinik für Hals-Nasen-Ohrenheilkunde. Andererseits liegen bei anderen seltenen Erkrankungen und Fehlbildungen weltweit deutlich unter 100 Fallberichte vor, was die Notwendigkeit einer Bündelung der verfügbaren Erfahrung zu Diagnostik und Therapie unterstreicht.
Die vorliegende Arbeit gibt eine Übersicht zu seltenen Erkrankungen der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis aus den übergeordneten Bereichen Erkrankungen / Syndrome des olfaktorischen Systems, Fehlbildungen der Nase und Nasennebenhöhlen, Belüftungs- und Funktionsstörungen sowie benigne und maligne Tumore. Die Einordnung sowie Angaben zu Diagnostik und Therapiemöglichkeiten wurden unter Berücksichtigung der aktuellen Literatur erstellt.
Schlüsselwörter
Seltene Erkrankungen - Nase - Nasenennebenhöhlen - vordere Schädelbasis - Fehlbildungen1. Einleitung
1.1 Definition und epidemiologische Gesichtspunkte seltener Erkrankungen
In der Europäischen Union wird eine Erkrankung als selten angesehen, wenn maximal 5 von 10 000 Personen von ihr betroffen sind [1]. In vielen Fällen stellen seltene Erkrankungen hinsichtlich der Stellung der richtigen Diagnose eine große interdisziplinäre Herausforderung dar. Innerhalb der EU sind ca. 6000 Erkrankungen als „selten“ gelistet. In Deutschland leiden ca. 4 Mio. Patienten an einer seltenen Erkrankung, europaweit wird die Zahl auf etwa 30 Mio. geschätzt. Aufgrund der Diversität der Erkrankungen und der jeweils nur geringen Anzahl betroffener Patienten ist die Forschung an Diagnostik und Therapie aus sozioökonomischen Gründen erschwert.
Anzahl und Definition der seltenen Erkrankungen sind aufgrund epidemiologischer Faktoren regional unterschiedlich. So können infektiöse Erkrankungen in Entwicklungsländern häufig auftreten, innerhalb Europas allerdings eine sehr geringe Inzidenz aufweisen.
1.2 Begriffsdefinition Inzidenz und Prävalenz
Inzidenz kennzeichnet die Anzahl neu auftretender Fälle einer Erkrankung in einer Population innerhalb eines definierten Zeitraums (meist 1 Jahr).
Prävalenz bezeichnet die gesamte Anzahl von Erkrankungen in einer Population zu einem bestimmten Zeitpunkt oder innerhalb eines definierten Zeitraums
Inzidenz = Neue Fälle/Gesamtbevölkerung
Prävalenz= Anzahlder Fälle/Gesamtbevölkerung x 100
Bei einigen Erkrankungen, die in der vorliegenden Arbeit beschrieben werden, existieren keine genauen Daten zur Inzidenz, da in der aktuellen Literatur nur sehr wenige Fallberichte beschrieben sind. Die Website http://www.orpha.net wurde 1997 in Frankreich ins Leben gerufen. Seit 2000 wird sie von der Europäischen Kommission gefördert und bietet Informationen zu vielen seltenen Erkrankungen. Die dort publizierten Informationen hinsichtlich Inzidenz und Prävalenz basieren auf gesammelten Originaldaten auf weltweiter oder europäischer Ebene oder extrapolierten Originaldaten, solange ein Founder-Effekt (Abweichung einer isolierten Population von einer Stammpopulation) für die Krankheit ausgeschlossen werden kann [2].
1.3 Nase, Nasennebenhöhlen und vordere Schädelbasis
Die menschliche Nase ist als Teil der oberen Atemwege u. a. für die Klimatisierung der eingeatmeten Luft verantwortlich. Die intakte Funktion der Mukosa beinhaltet einen Schleimfilm, der sich mithilfe der Zilien in konstanter Bewegung befindet. Diese Schicht stellt zum einen die Befeuchtung der eingeatmeten Luft sicher und stellt andererseits, gemeinsam mit dem Niesreflex, eine Barriere gegenüber eingeatmeten Fremdkörpern oder Erregern dar [3] [4]. Die gleichmäßige Verteilung der eingeatmeten Luft über eine möglichst große Fläche nasaler Schleimhaut ist für die intakte Funktion der Nase essenziell [4] [5] [6] [7]. Eine Vielzahl von Erkrankungen kann zu einer Beeinträchtigung oder dem Verlust der Klimatisierungs- und Schutzfunktion, aber auch des Riechvermögens führen [6] [8]. Neben typischen Erkrankungen wie der chronischen Rhinosinusitis mit Polyposis nasi (CRSwNP) oder den viralen Rhinitiden existieren auch einige seltene Erkrankungen, die zu Einschränkungen oder vollständigem Verlust dieser Funktionen führen und aufgrund der Ähnlichkeit der Symptome häufig spät diagnostiziert werden.
Nase, Nasennebenhöhlen und vordere Schädelbasis stellen aufgrund ihrer unmittelbaren Nachbarschaft eine anatomische Einheit dar, die hinsichtlich Erkrankungen in diesem Bereich nicht voneinander getrennt werden kann. So existieren nur wenige Erkrankungen, die dezidiert einen der genannten Bereiche betreffen. Insofern ist eine strikte Trennung der entzündlichen und tumorösen Erkrankungen sowie Fehlbildungen auf die Bereiche der Nasenhaupthöhle, der Nasennebenhöhlen oder der vorderen Schädelbasis nicht immer möglich, da viele Pathologien die Grenzen zwischen den einzelnen Unterbezirken überschreiten.
Sinunasale Tumore erfüllen mit einer Inzidenz von ca. 1–1,5:100 000 grundsätzlich die Voraussetzung einer seltenen Erkrankung [9] [10]. Diese malignen Neoplasien repräsentieren nur 3% aller Kopf-Hals-Karzinome und weniger als 1% aller malignen Erkrankungen des gesamten menschlichen Körpers [11] [12] [13] [14]. Insofern handelt es sich bei allen malignen Entitäten der Nase und Nasennebenhöhlen sowie der vorderen Schädelbasis per definitionem um seltene Erkrankungen.
Tumore der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis können nicht in jedem Fall nach anatomischer Region differenziert werden, da aufgrund der engen räumlichen Lagebeziehungen häufig bereits in niedrigen Stadien mehrere Regionen betroffen sind. Die TNM-Klassifikation der Karzinome der Nasenhaupthöhle und Nasennebenhöhlen ist in [Tab. 1] dargestellt.
|
T-Kategorien Nasenhaupthöhle und Siebbeinzellen |
|
|---|---|
|
T1 |
Tumor auf einen Unterbezirk der Nasenhöhle oder der Siebbeinzellen beschränkt, mit oder ohne Arrosion des Knochens |
|
T2 |
Tumor in zwei Unterbezirken eines Bezirks oder Ausbreitung auf einen Nachbarbezirk innerhalb des Nasen-Siebbeinzellen-Komplexes, mit oder ohne Arrosion des Knochens |
|
T3 |
Tumor breitet sich in die mediale Orbita oder den Orbitaboden aus oder in die Kieferhöhle, den Gaumen oder die Lamina cribrosa |
|
T4 |
Tumor infiltriert eine oder mehrere der folgenden Strukturen: |
|
T4a |
Inhalt der vorderen Orbita und/oder Haut von Nase oder Wange, minimale Ausbreitung in die vordere Schädelgrube, Processus pterygoidei, Keilbeinhöhle oder Stirnhöhle |
|
T4b |
Orbitaspitze und/oder Dura, Gehirn, mittlere Schädelgrube, Hirnnerven (ausgenommen den maxillären Ast des N. trigeminus [V2]), Nasopharynx, Clivus) |
2. Seltene Erkrankungen der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis
Seltene Erkrankungen der Nase, Nasennebenhöhlen und Schädelbasis können in folgende Kategorien eingeteilt werden:
-
Erkrankungen/Syndrome des olfaktorischen Systems
-
Fehlbildungen
-
Belüftungs- und Funktionsstörungen
-
Benigne und maligne Tumore
-
Entzündliche/granulomatöse Erkrankungen
In einem Referat aus dem Jahr 2015 wurden bereits seltene rhinologische Erkrankungen mit Fokus auf eine granulomatöse Genese durch Martin Laudien dargelegt [15], weshalb die vorliegende Arbeit auf die ersten vier der genannten Aspekte fokussiert. Hierbei werden die wichtigsten seltenen Erkrankungen des olfaktorischen Systems, Fehlbildungen, Tumorerkrankungen und Funktionsstörungen der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis unter Berücksichtigung der aktuellen Literatur dargestellt.
2.1 Erkrankungen/Syndrome des olfaktorischen Systems
Die Genese von Störungen des olfaktorischen Systems ist sehr variabel. Mögliche Ursachen liegen in rhinologischen Erkrankungen, Traumata, Neoplasien und kongenitalen Störungen oder sie werden als idiopathisch bezeichnet [16] [17]. Zudem existieren viele virale Erkrankungen, die mit einer temporären und permanenten Hyp- bzw. Anosmie einhergehen können. Aufgrund der Themenstellung fokussiert die vorliegende Arbeit nicht auf alle seltenen Erkrankungen, die auch das olfaktorische System beeinträchtigen können, sondern auf jene, die primär das Riechen betreffen. Hierbei handelt es sich um folgende Syndrome bzw. Fehlbildungen:
-
Isolierte kongenitale Anosmie
-
Kallmann-Syndrom
-
Neuroektodermales Syndrom Typ Johnson
2.1.1 Isolierte kongenitale Anosmie
Die isolierte kongenitale Anosmie ist extrem selten. Weltweit wurden bislang 15 Fälle einer isolierten kongenitalen Anosmie beschrieben [18]. Bei der isolierten kongenitalen Anosmie liegt eine seit Geburt bestehende Anosmie vor. Die Ursache ist eine Entwicklungsstörung des Bulbus olfactorius, die uni- oder bilateral vorliegen kann [19]. Eine weitere Genese scheint in dem Ersatz des olfaktorischen Epithels durch respiratorisches Epithel, welches die Nasenhaupthöhle auskleidet, zu liegen. Ursächlich ist eine autosomal dominante Störung mit unvollständiger Penetranz. Es existiert keine kausale Therapie der Fehlbildung.
2.1.2 Kallmann-Syndrom
Das Kallmann-Syndrom beschreibt eine erbliche Entwicklungsstörung, in deren Rahmen es zu einem kongenitalen hypogonadotropen Hypogonadismus aufgrund einer Gonadotropin-Releasing Hormon (GnRH)-Mangels kommt. Zusätzlich ist bei betroffenen Patienten eine Hyposmie oder Anosmie (bei Hypoplasie oder Aplasie des Bulbus olfactorius) zu beobachten, die auf eine unterbrochene embryonale Migration der GnRH-synthetisierenden Neuronen vom Riechepithel in die Hypothalamusregion basiert. Die Vererbung erfolgt X-chromosomal rezessiv [20]. Die Prävalenz des Kallmann-Syndroms wird auf 3,75:100 000 geschätzt [2]. Therapien zielen auf die Induktion der Pubertät und später der Fertilität ab. Während in diesem Bereich gute Therapieerfolge erzielt werden können, ist eine Behandlung der Hyposmie oder Anosmie nicht möglich.
2.1.3 Neuroektodermales Syndrom Typ Johnson
Das Neuroektodermale Syndrom Typ Johnson beinhaltet die Symptome Alopezie, Anosmie oder Hyposmie, Schallleitungs-Schwerhörigkeit, malformierte Ohren, Mikrotie und/oder Atresie des äußeren Gehörganges und hypogonadotropen Hypogonadismus. Es wird nach den Erstbeschreibern auch Johnson-McMillin Syndrom bezeichnet [21] [22]. Die Vererbung erfolgt autosomal dominant, allerdings ist die genaue Ätiologie nicht bekannt. Vermutet wird ein embryologischer Defekt bei der Ausdifferenzierung der Neuralleiste der Kopfregion.
Die Prävalenz des Syndroms wird auf deutlich unter 1:1.000 000 geschätzt. Eine Therapie der mit dem Syndrom assoziierten Hyposmie bzw. Anosmie ist nicht existent.
2.2 Fehlbildungen der Nase und Nasennebenhöhlen
In der aktuellen Literatur existieren mehr als 300 Syndrome mit Beteiligung der Nase. Bei vielen dieser Syndrome kann auch die Entwicklung der Nase beeinträchtigt sein, allerdings ist diese Pathologie im Gesamtaspekt der vielen, im Rahmen des Syndroms vorkommenden Pathologien als untergeordnet zu beurteilen. Der Übersichtlichkeit halber werden im folgenden Teil Pathologien dargestellt, die primär die Nase und je nach Ausprägung auch die Nasennebenhöhlen und vordere Schädelbasis betreffen. Hierbei handelt es sich um folgende Fehlbildungen:
-
Arrhinie/Hemirhinie
-
Bifide Nase
-
Kraniorhinie
-
Kraniofaziale Gesichtsspalten und paramediane Nasenspalten
-
Duplikationsanomalien der Nase
-
Proboscis lateralis
-
Nasenrückenfistel
2.2.1 Arrhinie/Hemirhinie
Die kongenitale Arrhinie stellt eine extrem seltene Fehlbildung dar, bei der die äußere Nase nicht oder nur sehr rudimentär angelegt ist ([Abb. 1]) und deren Pathogenese noch nicht vollständig analysiert werden konnte. Es wird vermutet, dass eine Entwicklungsstörung der paarigen Nasenplakoden zwischen der 3. und 10. Gestationswoche eine Rolle in der Genese spielt. Die vorzeitige Fusion der medianen Nasenfortsätze, eine fehlende Resorption der nasalen Epithelpfropfen sowie eine abnorme Wanderung von Zellen der Neuralleiste werden als weitere Entstehungsmechanismen diskutiert. Eine Arrhinie kann als eigenständige Fehlbildung oder im Rahmen von Syndromen wie dem Treacher-Collins Syndrom, das eine oto-mandibuläre Dysplasie mit verschiedenen Defekten an Kopf und Hals beinhaltet und dem „Arhinie-Mikrophthalmie-Syndrom Bosma“ bzw. „Bosma-Henkin-Christiansen-Syndrom“, einer Kombination aus Arrhinie, Choanalatresie und Mikrophthalmie, auftreten.

Weltweit wurden ca. 20 Fälle beschrieben [2]. Noch seltener sind Fälle von kongenitaler einseitiger Arrhinie (Hemirhinie, fehlende Nasenhälfte ([Abb. 2])). In einigen Fällen wird das vollständige Fehlen einer Nasenanlage mit palpatorisch festem Untergrund beschrieben ([Abb. 1]), andere Fälle zeigen eine rudimentäre Nasenanlage als blind endender Buckel oder Grübchen [23].

Die meisten berichteten Fälle einer Arrhinie sind sporadisch aufgetreten und zeigen einen unauffälligen Karyotyp [24] [25]. Die Überlebensrate von Patienten mit dieser Fehlbildung ist niedrig, da es in ihrem Rahmen zu schweren oberen Atemwegsobstruktionen, Atemnot und Infektionen der Atemwege sowie Mangelernährung kommen kann [26]. Die chirurgische Therapie ist Mittel der Wahl, allerdings existieren nur wenig Daten hinsichtlich der Technik und des besten Zeitpunktes für eine Operation. Eine Tracheostomie sollte initial erfolgen, um die Atmung sicherzustellen. Allgemein wird empfohlen, die rekonstruktive Operation spätestens bis zum Vorschulalter zu planen.
2.2.2 Bifide Nase
Bei der bifiden Nase handelt es sich um eine seltene angeborene Missbildung mit vermutlich autosomal-dominantem oder rezessivem Erbgang. Kennzeichen ist eine Nasenspalte, deren Ausprägung sehr variabel ist. Diese reicht von einer wenig auffälligen Furche am Nasensteg oder der Nasenspitze ([Abb. 3]) bis zur vollständigen Spaltung der darunter liegenden Knochen und Knorpel, was zur Ausbildung zweier Halbnasen führen kann. Der Atemweg ist im Rahmen dieser Fehlbildung meist adäquat ausgebildet. Eine bifide Nase kann als milde Form einer frontonasalen Dysplasie gesehen werden [27], aber auch andere Fehlbildungen wie Hypertelorismus und Mittellinienspalten der Lippe treten in Zusammenhang mit der Nasenfehlbildung auf [28].

Die Ursache der frontonasalen Dysplasie ist unbekannt. Die vermutete Ätiologie ist ein Fehler bei der Entwicklung der Nasenkapsel, in dessen Verlauf zwischen der 4. und 6. Woche der Embryogenese die Migration des olfaktorischen Epithels in die Nasenkapsel aufgehalten wird. Diese bildet sich nicht vollständig aus und das primitive Hirngewebe füllt den Raum zwischen dem dehiszenten Nasenrücken aus [29].
Eine CT oder MRT Bildgebung ist vor einer operativen Therapie dringend erforderlich, da auch milde Formen der frontonasalen Dysplasie mit intrakraniellen Anomalien einhergehen können.
Operativ wird bei der milden Verlaufsform einer bifiden Nase i.d.R. eine offene Rhinoplastik durchgeführt [27], was ein übersichtlicheres Operationsfeld ermöglicht und die Gefäßversorgung der Haut und des Weichteilmantels der Nase erhält. Es wird angenommen, dass die Columellainzision sowie die Anhebung der Nasenhaut, die bei der offenen Technik angewandt werden, das Wachstum der kindlichen Nase nicht beeinflusst [30].
2.2.3 Kraniorhinie
Merkmale einer Kraniorhinie sind Brachyzephalie, fliehende Stirn, eine sklerotische Schädelbasis, allerdings ohne Kraniosynostosen. Der Ductus nasolacrimalis ist nicht ausgebildet. Die Nase ist breit konfiguriert, die Alae nasi wirken aufgebläht und antevertiert. Es zeigt sich ein nasaler Hirsutismus und bilateral symmetrische, kugelförmige, zystenartige Strukturen mit kleinen Fisteln direkt unterhalb der Nase. Ein Hypertelorismus kann zusätzlich vorhanden sein. Die Vererbung ist wahrscheinlich autosomal dominant, allerdings legen Fälle mit konsanguinen Eltern nahe, dass eine pseudodominante autosomal rezessive Übertragung nicht völlig ausgeschlossen werden kann [31] [32]. Weltweit wurden vier betroffene Familien beschrieben [2].
2.2.4 Kraniofaziale Gesichtsspalten und paramediane Nasenspalte
Kraniofaziale Gesichtsspalten sind äußerst seltene Fehlbildungen der Embryogenese. Primäre oder echte Spaltbildungen treten zwischen der 4. und 8. Gestationswoche auf, weil die Fusion zwischen den verschiedenen Gesichtsprozessen nicht vollständig abgeschlossen wurde. Sekundäre oder Pseudospalten treten später auf. Sie betreffen die mesenchymale Differenzierung und können als Dysplasie bezeichnet werden. In beiden Situationen ist das zukünftige Wachstumspotenzial im Vergleich zum übrigen Gesicht vermindert. Die Inzidenz kraniofazialer Gesichtsspalten wird mit 1,4–4,9:100 000 angegeben [33].
Von der Spaltbildung kann Hirngewebe, Weichgewebe und Knochen betroffen sein. Knöcherne Fehlbildungen treten an der Stirn, Orbita, Sinus ethmoidalis, Maxilla und Gaumen auf. Meningozelen und Meningoenzephalozelen können bei intrakranieller Beteiligung vorkommen.
Mediane und paramediane Gesichtsspalten sind häufig mit Hypertelorismus, anteriorer oder basaler Enzephalozele, Lageanomalien des Oberkiefers und Nasendeformitäten assoziiert ([Abb. 4]). Es können auch Weichteilmissbildungen wie Lippen- und Gaumenspalten und Lidkolobome auftreten [33].

Die paramediane Nasenspalte stellt einen seltenen Entwicklungsdefekt während der Embryogenese dar, der durch ein einseitiges oder beidseitiges Kolobom der Nase gekennzeichnet ist ([Abb. 5]). Sie ist eine milde Form einer kraniofazialen Gesichtsspalte. Die Fehlbildung kann in Form einer kleinen Kerbe, die zu einer geringfügigen Abweichung der Nasenscheidewand führt, bis hin zu Nasenspalten unterschiedlicher Größe, die mit kleinen Zysten der Nasennebenhöhlen in der Nasenmittellinie assoziiert sein können, auftreten. Paramediane Nasenspalten treten isoliert oder in Verbindung mit Lippenspalten und/oder anderen kraniofazialen Anomalien (z. B. Hypertelorismus, Verbreiterung der Nasenwurzel, Mittellinienspalte) auf. Nasenrücken und Nasenspitze sind in der Regel gut erhalten [34].

2.2.5 Duplikationsanomalien der Nase: Polyrhinie und akzessorisches Nasenloch
Duplikationsanomalien der Nase beinhalten die Polyrhinie („doppelte Nase“) und das akzessorische Nasenloch. Beide Nasendeformitäten treten extrem selten auf (entsprechend Orphanet wird die Inzidenz auf unter 1:1.000 000 geschätzt). In der Literatur sind insgesamt 8 Fälle beschrieben, von denen 4 eine Polyrhinie und 3 ein isoliertes akzessorisches Nasenloch sowie ein Patient ein akzessorisches Nasenloch in Verbindung mit einer Lippenspalte aufwiesen [35].
Die Polyrhinie ist eine angeborene Fehlbildung, bei der es zu einer vollständigen Duplikation der Nase kommt. Alle publizierten Fälle waren sporadisch. Es wird vermutet, dass der Fehlbildung ein embryonaler Entwicklungsdefekt mit Duplikation des medialen Nasenfortsatzes zugrunde liegt [36].
Das akzessorische Nasenloch ist eine ebenfalls äußerst seltene angeborene Fehlbildung, die durch das Vorhandensein eines oder mehrerer akzessorischer Nasenlöcher mit oder ohne akzessorischen Knorpel gekennzeichnet ist. Die akzessorischen Löcher sind medial, oberhalb, unterhalb oder lateral der anderen Nasenlöcher lokalisiert. Im Gegensatz zur Polyrhinie besteht keine Duplikation des Nasenseptums. Das akzessorische Nasenloch ist häufig mit anderen Fehlbildungen der Kopf-/Halsregion assoziiert [37] [38].
2.2.6 Proboscis lateralis
Der Proboscis lateralis ist eine äußerst seltene Fehlbildung, die 1861 zum ersten Mal beschrieben wurde. Die fehlgebildete Seite der Nase zeigt eine schlauchartige, rudimentäre Nasenanlage, die an jedem beliebigen Punkt entlang der embryologischen Fusionslinie zwischen der anterioren Maxilla und dem Processus frontonasalis ansetzen kann. In den meisten Fällen ist der Ansatz dieses Rudiments am medialen Anteil des Orbitadaches lokalisiert [39].
Der genaue embryologische Mechanismus, der für die Entwicklung des Proboscis lateralis verantwortlich ist, konnte bislang nicht analysiert werden. Zu den populären Theorien gehören die unvollkommene Verschmelzung der lateralen Nasen- und Kieferfortsätze und die irreguläre Verschmelzung des Kieferfortsatzes der betroffenen Seite mit dem medialen Nasenfortsatz [40] [41] [42].
Entsprechend den existierenden Daten wird empfohlen, die chirurgische Therapie in der frühen Kindheit zu beginnen, um mögliche psychosoziale Konsequenzen zu vermeiden. Die abschließende ästhetische Rekonstruktion der Nase sollte allerdings erst in der späteren Jugend erfolgen, wenn das Wachstum des Nasengerüsts abgeschlossen ist.
Es existieren verschiedene chirurgische Techniken zur initialen Korrektur des Proboscis lateralis. Eine häufig angeführte Technik beschreibt die Desepithelisierung des mittleren bis distalen Anteils des rudimentären Schlauches, der dann in die eröffnete und fehlgebildete ipsilaterale Nasenwand eingearbeitet wird [39].
2.2.7 Nasenrückenfistel
Die Nasenrückenfistel ist eine seltene Fehlbildung, die sich durch das Vorhandensein einer Dermoidzyste auf dem Nasenrücken definiert. Die Inzidenz liegt bei 1:20 000–1:40 000 [43]. Klinisch präsentiert sich eine fest palpable, langsam wachsende Raumforderung, die Haut und dermale Elemente, wie Haarfollikel und Talgdrüsen enthält. Es kann zu intermittierender oder chronischer Sekretion von Talg und seröser Flüssigkeit sowie zu lokalen Infektionen kommen. In einzelnen Fällen besteht eine Verbindung nach intrakraniell, weshalb anamnestisch auf meningitische Beschwerden und Krampfanfälle fokussiert werden sollte. In sehr seltenen Fällen kann es zu intrakraniellen Abszessformationen kommen. Therapie der Wahl ist die vollständige Exzision des Befundes, je nach Ausdehnung in Kooperation mit der Neurochirurgie. Bei vollständiger Resektion sind Rezidive selten [44].
2.3 Belüftungs- und Funktionsstörungen
2.3.1 Silent Sinus Syndrom
Das Silent Sinus Syndrom bezeichnet eine i.d.R. einseitige Erkrankung des Sinus maxillaris, die mit einem verringerten Volumen, einer Absenkung des Orbitabodens und einem damit assoziierten Bulbustiefstand (Hypoglobus) assoziiert ist [45]. Bereits 1964 wurden von Montgomery 2 Fälle eines Enophthalmus bei maxillären Mukozelen beschrieben [46], der Begriff Silent Sinus Syndrom entstand im Rahmen der Beschreibung einer Fallserie von 19 Patienten mit Enophthalmus und einem einseitigen „Kollaps“ des Sinus maxillaris [47]. In extrem seltenen Fällen ist das Siebbein oder die Stirnhöhle von der Erkrankung betroffen [48] [49]. Weltweit wurden bisher 98 Fälle beschrieben [2].
Betroffene Patienten sind weitestgehend frei von sinunasalen Beschwerden, können aber über gelegentliches Druckgefühl der betroffenen Seite des Oberkiefers berichten [50] [51]. Trotz der durch den Enophthalmus und Hypoglobus veränderten Sehachse kommt es wegen der nur langsam fortschreitenden Symptomatik nur selten zu Visusstörungen.
Als wahrscheinlichste Ursache für die Entstehung eines Silent Sinus Syndroms wird eine Obstruktion am Ostium der betroffenen Nebenhöhle vermutet. Es kommt zu einer konsekutiven Unterdruckbildung, eingezogenen knöchernen Wänden und nachfolgenden Sekretretention [52] [53] [54] [55] [56]. Die ursprüngliche These, es handele sich um eine Kieferhöhlen-Hypoplasie, konnte durch Studien widerlegt werden, bei denen Computertomografien aus der Zeit vor der Erkrankung analysiert wurden, auf denen ein normal konfigurierter Sinus maxillaris ersichtlich war [54] [57] [58]. Auch eine chronische Rhinosinusitis wird als Ursache diskutiert [56].
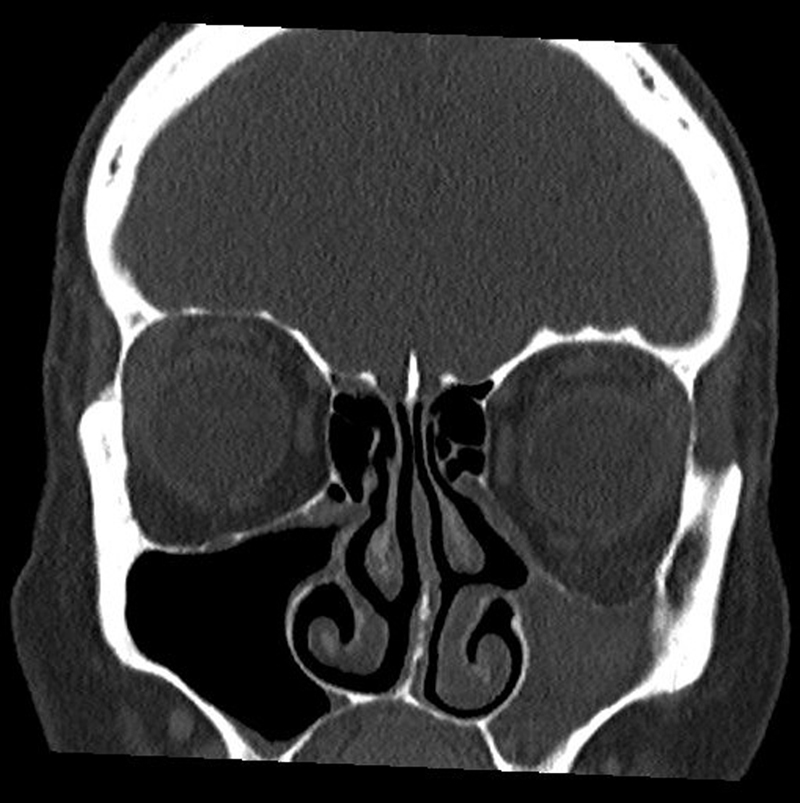
Bei der Endoskopie zeigt sich ein stark retrahierter, ggf. atelektatischer Processus uncinatus. Computertomografisch lässt sich ein hypoplastisch anmutender Sinus maxillaris mit gleichmäßiger Einziehung aller Wände darstellen. Das Lumen des Sinus kann frei oder Sekret-verlegt sein [59]. In koronarer Schichtung lässt sich in der Computertomografie der nach kaudal eingezogene Orbitaboden darstellen, der zu einer konsekutiven Verlagerung des Orbitainhalts führt ([Abb. 6]). Magnetresonanztomografisch lässt sich ein hyperintenses Signal in der T2-Wichtung bei homogener Abbildung des Lumens darstellen [51] [59] [60] [61].

Bei entsprechenden Beschwerden ist eine Kieferhöhlen-Fensterung Typ 2–3 [56] [62] [63] [64] zur Wiederherstellung der Belüftung möglich. Nach Eröffnung des Lumens zeigt sich normale bis leicht hypertrophe Schleimhaut und je nach Befund mukozelenartig konfiguriertes Sekret, welches abgesaugt werden kann.
Im Falle von Visusstörungen ist die Korrektur des Orbitabodens Gegenstand zahlreicher Diskussionen. Zur Rekonstruktion bzw. Anhebung des Orbitabodens können alloplastische oder autologe Materialen verwendet werden [55] [65]. Mehrere Fallberichte zeigten allerdings eine Rückbildung des Enophthalmus und Hypoglobus [52] [57] [65] [66], weshalb eine operative Therapie frühestens ein Jahr nach Wiederherstellung der Kieferhöhlen-Belüftung erfolgen sollte, um spontane Remissionen abzuwarten [62] [65] [67].
2.3.2 Hypersinus
Der Terminus Hypersinus bezeichnet eine Nasennebenhöhle, deren Ausdehnung die normalen Begrenzungen deutlich überschreitet und deren knöcherne Begrenzungen und Belüftungssituation normal konfiguriert sind [68]. Die Ausdehnung der betroffenen Nebenhöhle erstreckt sich innerhalb der Schädelknochen-Begrenzungen, ohne diese – wie im Falle des Pneumosinus dilatans oder der Pneumozele – zu verdrängen bzw. zu verlagern. Insofern existieren bei betroffenen Patienten keine kosmetischen oder funktionellen Probleme. In [Abb. 7] ist die starke Pneumatisation des Sinus frontalis beidseits gut zu erkennen. In sagittaler Rekonstruktion zeigt sich der weit nach kranial reichende Sinus frontalis ebenfalls, allerdings ohne die Kontur der Stirn stark zu beeinträchtigen.

Der Hypersinus verursacht keine Beschwerden, weshalb diese anatomische Variante nicht als Pathologie gezählt wird. Trotz einer bereits 1987 festgelegten Nomenklatur [69] werden die Begriffe Hypersinus, Pneumosinus dilatans und Pneumozele bisweilen unscharf oder sogar synonym verwendet, weshalb die Abgrenzung des Hypersinus in diesem Zusammenhang aufgeführt wird.
2.3.3 Pneumosinus dilatans
Der Pneumosinus dilatans stellt eine massive Ausweitung meist einer Nasennebenhöhle dar und manifestiert sich in den meisten Fällen an der Stirnhöhle. Auch die Siebbeinzellen und die Keilbein- sowie Kieferhöhlen können von der außergewöhnlich starken Pneumatisation betroffen sein [56]. Vorwölbungen des Stirnbeins oder intrakranielle, ethmoidale und orbitale Ausdehnung können auftreten [69]. Die Erkrankung ist nicht zwingend mit Beschwerden verknüpft, kann aber in einigen Fällen lokales Druckgefühl und Cephalgien verursachen. Während der Befall der Stirnhöhle zu kosmetischen Beeinträchtigungen für den Patienten führen kann, existieren Fallberichte mit ophthalmologischen Komplikationen bei Lokalisation im Siebbein oder der Keilbeinhöhle [70] [71] [72] durch eine Kompression des Nervus opticus.
Die weltweit berichtete Fallzahl wird mit 134 angegeben [73] [74]. Die Ätiologie der Erkrankung ist unbekannt. Als mögliche Ursachen werden spontan drainierte Mukozelen, eine Infektion mit gasbildenden Mikroorganismen, eine genetische Prädisposition oder fibro-ossäre Dysregulation sowie hormonelle Dysregulationen diskutiert [74]. Die derzeit wahrscheinlichste Theorie ist ein Ventilmechanismus der Abflusswege der Nebenhöhlen, der aufgrund des gesteigerten Drucks zu einer langsamen, gleichmäßigen Ausdehnung des Sinus führt und spontan drainierte Mukozelen diskutiert. Publikationen der letzten 20 Jahre geben zudem Hinweise auf eine Assoziation mit Meningeomen und Arachnoidalzysten [73] [75] [76] [77].
Bei der Nasenendoskopie zeigt sich ein meist unauffälliger Befund des mittleren Nasengangs und der betroffenen Ostien, sofern diese einsehbar sind. Computertomografisch ist die betroffene Nasennebenhöhle (meist) gleichmäßig vorgewölbt und i.d.R. ohne Ausdünnung des begrenzenden Knochens darstellbar. [Abb. 8] zeigt eine koronare Computertomografie eines Patienten mit Pneumosinus dilatans. In sagittaler Rekonstruktion ist die Vorwölbung der Stirnhöhlenvorderwand sowie der hierdurch veränderte nasofrontale Winkel zu erkennen.

Es existiert keine kurative Therapie. Unter der Annahme eines Ventilmechanismus kann eine funktionelle Nasennebenhöhlen-Operation mit Erweiterung des Ostiums und damit Beseitigung der Stenose in Betracht gezogen werden. Bei Patienten mit kosmetischer Beeinträchtigung im Rahmen eines Pneumosinus dilatans frontalis wurden Techniken zur operativen Modellierung der Stirnhöhlenvorderwand von Draf et al. beschrieben [78]. Bei Visusstörungen kann in Abhängigkeit der Beschwerden und der Lokalisation der Pathologie die Dekompression des Nervus opticus in Erwägung gezogen werden. Auch ein Pneumosinus dilatans der Kieferhöhle kann sich entwickeln und zeigt sich primär in einer äußeren Deformierung [56].
2.3.4 Pneumozele
Pneumozelen sind Ausdehnungen einer Nasennebenhöhle über die normalen Grenzen hinaus. Im Gegensatz zum Pneumosinus dilatans finden sich Irregularitäten der knöchernen Begrenzung der betroffenen Nebenhöhle mit Ausdünnungen und teilweisem Integritätsverlust [69] [79]. Die Symptome sind ähnlich wie bei einem Pneumosinus dilatans. Bei Lokalisation im Sinus maxillaris, frontalis und ethmoidalis kann es zu Verlagerung des Orbitainhalts mit konsekutiver Protrusio bulbi kommen [80] [81] [82]. Ein Fallbericht schildert einen temporären Visusverlust im Rahmen einer Pneumocele des Sinus sphenoidalis [83] [84].
Als Ursache für die Entwicklung einer Pneumozele wird ein Ventilmechanismus im Bereich des Ostiums angenommen, der eine rasche Angleichung von Druckdifferenzen zwischen der Nasenhaupthöhle und der betroffenen Nebenhöhle verhindert.
Im Falle einer Protrusio bulbi kann die Entlastung der Orbita mit Resektion der Lamina papyracea erfolgen. Bei einer Kompression des Nervus opticus im Bereich des Sinus sphenoidalis aufgrund von nicht ausgeglichenen Druckdifferenzen der Nasenhaupthöhle und des Sinus sphenoidalis ist die operative Wiederherstellung der Belüftung des Sinus sphenoidalis Therapie der Wahl. In o.g. Fallbericht wurde eine Resektion eines Polypen durchgeführt, der das Ostium verlegte.
2.3.5 Organisiertes Hämatom
Das sinunasale organisierte Hämatom ist eine seltene, gutartige Erkrankung. Es kommt zu wiederholten Einblutungen, die möglicherweise durch ein sehr kleines Ostium und/oder eine nicht ausreichende mukoziliäre Clearance des betroffenen Sinus raumfordernden Charakter entwickeln. Im weiteren Verlauf kommt es zu Fibrosierungen und Neovaskularisation. Aufgrund des expansiven Wachstums kann es zu Destruktionen umgebender Strukturen kommen, weshalb bildmorphologische Aspekte malignen Wachstums oder einer Pathologie mit lokal aggressiver Expansion ähnlich einem invertierten Papillom oder einer Pilzsinusitis entstehen können [85] [86]. Diverse Studien haben die Charakteristika der Erkrankung analysiert, allerdings ohne genauere Daten hinsichtlich der Inzidenz zu liefern [85] [86] [87].
Primäre Beschwerden sind häufig rezidivierende Epistaxis, Schmerzen, Druckgefühl im Gesichtsbereich und gelegentlich Hypästhesien im Versorgungsgebiet des N. infraorbitalis [85] [86] [87] [88].
CT- und MRT-grafisch zeigen sich mit expansiven Gewebsmassen angefüllte Sinus, häufig mit Ausdehnung in die ipsilaterale Nasenhaupthöhle [87]. Lokal aggressives Wachstum kann zu Expansion in den Sinus ethmoidalis, die Orbita, die Fossa pterygopalatina und die Fossa infratemporalis, die Wange und sogar den Hartgaumen führen. In der Computertomografie stellen sich die Areale heterogen und mit unregelmäßig fleckiger Kontrastmittelaufnahme dar. Auch Kalzifikationen können auftreten. Magnetresonanztomografisch zeigen sich vergleichbare Kontrastmittelanreicherungen in der T1 Wichtung mit eher hypointensem Randsaum.
Histopathologisch zeigen sich Einblutungen mit frischeren und älteren Arealen, extensiv dilatierte Gefäße, amyloides Material mit irregulär konfigurierten Gefäßen, Zonen mit deutlicher Neovaskularisation sowie Hämosiderinablagerungen und Fibrosen [87].
Die explorative Endoskopie in Narkose wird zur Diagnosesicherung und Therapie empfohlen. Über einen endonasalen Zugang sollte das Hämatom vollständig entfernt werden. Im Rahmen der Ausräumung kommt es häufig zu stärkeren diffusen Blutungen. Um einer weiteren Abkapselung des Befundes vorzubeugen wird empfohlen, ausreichend weite Zugänge zu dem betroffenen Sinus anzulegen. Rezidive sind in der derzeit bekannten Literatur äußerst selten beschrieben [85] [86] [87] [88].
Die zu erwartenden endoskopischen und radiologischen Befunde bei Silent-Sinus-Syndrom, Organisiertem Hämatom und Pneumosinus dilatans sind in [Tab. 2] aufgelistet.
|
Erkrankung |
Endoskopischer Befund |
Radiologischer Befund |
|---|---|---|
|
Silent-Sinus-Syndrom |
Erweiterung des mittleren Nasengangs mit Lateralisation des Proc. uncinatus |
CT:
|
|
Organisiertes Hämatom |
Gewebsvermehrung im mittleren Nasengang und in der Nasenhaupthöhle, Fibrin, Granulationen. Z. T. polypöse Schleimhautschwellung und Vorwölbung der lateralen Nasenwand |
CT:
MRT:
|
|
Pneumosinus dilatans |
Unauffälliger Befund |
CT: Erweiterung des Sinus maxillaris über die natürlichen Grenzen hinaus ohne Ausdünnung der knöchernenWände |
2.3.6 Young-Syndrom
Erstmals beschrieben wurde das Young-Syndrom von dem Urologen David Young, dem aufgefallen war, dass 54% der Patienten mit obstruktiver Azoospermie Lungendefekte aufwiesen [89]. Im Jahr 1978 wurden sinunasale Beschwerden in die Symptomatik aufgenommen. Seither ist das Krankheitsbild als Trias aus obstruktiver Azoospermie, chronischer Rhinosinusitis und Bronchiektasien oder chronischen Bronchitiden definiert [90] [91].
Hinsichtlich der Inzidenz des Young-Syndroms existieren keine verlässlichen Daten. Eine Quecksilber-Exposition scheint mit dem Auftreten der Erkrankung assoziiert zu sein. Untermauert wird dies durch die nach der Erstbeschreibung beschriebene hohe Inzidenz von bis zu 1 von 500 Männern auf heute nur noch wenige Fallberichte, was dem generellen Verschwinden von Quecksilber aus Industrie und Medizin geschuldet sein dürfte [92]. Eine positive Familienanamnese ist laut aktuellem Stand keine Prädisposition für das Auftreten der Erkrankung.
Das Young-Syndrom betrifft junge Männer. Primärer Grund für das Aufsuchen ärztlicher Hilfe ist in den meisten Fällen die Infertilität, nur selten chronisch sinunasale oder pulmonale Beschwerden. Die chronisch sinunasalen Beschwerden verschwinden mit dem Abschluss der Adoleszenz wohingegen die pulmonalen Beschwerden persistieren [93] [94] [95].
Die mukoziliäre Clearance ist bei betroffenen Patienten deutlich verlängert, was allerdings kein spezifisches diagnostisches Kriterium darstellt [96]. Initial vermutete Strukturdefizite der Dynein-Arme innerhalb der Zilien konnten nicht als Ursache bestimmt werden. Stattdessen geht man heute von einer veränderten Konsistenz des nasalen Schleimfilms aus, die für die Beschwerden der Patienten verantwortlich ist [91].
Differenzialdiagnostisch muss an eine Mukoviszidose, eine primär ziliäre Dyskinesie und das Kartagener Syndrom gedacht werden.
Wang et al. untersuchten ein Kollektiv von 33 Patienten mit obstruktiver Azoospermie und beschrieben 4 Patienten mit einer dokumentierten Anamnese einer chronischen Rhinosinusitis, einer auffälligen Bildgebung der Nasennebenhöhlen, einer positiven Familienanamnese sowie einer Medikation, die die mukoziliäre Clearance beeinträchtigen kann. Da die Anzahl der in der Literatur dargelegten Fälle seit der Erstbeschreibung massiv gesunken ist und aufgrund der lange inkonsistent verwendeten Definition einer chronischen Rhinosinusitis wurde die Existenz des Young-Syndroms 2009 von Arya et al. sogar angezweifelt [91].
2.3.7 Primäre ziliäre Dyskinesie
Die primäre ziliäre Dyskinesie ist eine Störung der Struktur und Funktion der beweglichen Zilien der Schleimhaut der Nasenhaupthöhle und der Nasennebenhöhlen, die zu chronischen sinunasalen und pulmonalen Beschwerden führt. Die primäre ziliäre Dyskinesie äußert sich typischerweise in Atembeschwerden bei Säuglingen, frühem ganzjährigem Husten und einer Nasenatmungsbehinderung. Aufgrund der fehlenden Zilienfunktion kommt es zu einer Stase des Schleimhautfilms in der Nase und den Nasennebenhöhlen, die bei betroffenen Patienten zu einer purulenten Sekretion führt. Die korrekte Diagnosestellung ist eine Herausforderung, da eine Vielzahl von Erkrankungen existiert, die zu ähnlichen Symptomen führen. Das Kartagener-Syndrom ist eine Trias aus chronischer Rhinosinusitis, Bronchiektasien und dem Vorliegen eines Situs inversus als Folge einer ziliären Dyskinesie [97].
Ursächlich ist eine genetische Störung, die zu einer gestörten Ultrastruktur der Zilien der Nasenschleimhaut und damit deren Funktionslosigkeit führt. Derzeit sind 33 Gene bekannt, die mit der Ausbildung einer primären ziliären Dyskinesie assoziiert sind und deren Mehrheit einem autosomal rezessivem Erbgang folgt [97]. Die Prävalenz liegt bei 1:15 000 Lebendgeburten
Bei der primären ziliären Dyskinesie führen Mutationen in Genen, die für axonemale Strukturen kodieren, zu funktionsgestörten Zilien. Defekte bei der primären ziliären Dyskinesie können äußere Dyneinarmdefekte, innere Dyneinarmdefekte, zentrale mikrotubuläre Anomalien, radiale Speichendefekte und äußere ultrastrukturelle Anomalien beinhalten. Auch der Spermienschwanz und die Fimbrien der Eileiter besitzen bewegliche Zilien, weshalb Infertilität bei betroffenen Männern und Frauen auftreten kann. Anomalien der Anatomie sind möglich, da der Defekt der beweglichen Flimmerhärchen während der Embryogenese zu einer abnormalen thorakoabdominalen Ausrichtung führt. Ein Situs inversus tritt bei 50% der Fälle von primärer ziliärer Dyskinesie auf, da die normale Bewegung der Zilien gestört ist und die viszerale Rotation hierdurch ein Zufallsereignis wird [97] [98] [99].
Die Diagnose wird durch eine Kombination aus Symptomen und den Ergebnissen einer Nasen- oder Bronchialbürstenbiopsie zum Nachweis einer gestörten ziliären Ultrastruktur und Ziliarbeweglichkeit gestellt. Die Analyse der Nasenschleimhaut mit Hochgeschwindigkeits-Videomikroskopie zur Beurteilung der Ziliarmotilität ist sehr sensitiv und spezifisch.
Bei Patienten über 5 Jahren sind nasale Stickstoffmonoxidmessungen sensitiv und können die Diagnosestellung erleichtern. Der Gehalt des durch die Mukosa produzierten Stickstoffmonoxids ist bei Patienten mit primärer ziliärer Dyskinesie im Vergleich zu gesunden Patienten deutlich erniedrigt. Aufgrund der teilweise ähnlichen Symptome sind ein Schweiß-Chlorid-Test und ggf. genetische Tests sinnvoll, um eine Mukoviszidose ausschließen zu können [97] [100] [101] [102].
Es existieren keine großen, langfristig angelegten randomisierten Studien zur Therapie der primären ziliären Dyskinesie, weshalb viele Aspekte der Versorgung auf empirischen Empfehlungen anderer Lungenerkrankungen mit ähnlichen Pathologien basieren. Trotz des Versuches, einen europäischen Konsensus aus den Erfahrungen großer spezialisierter Zentren zu erzielen, existieren große Unterschiede in den Ansätzen, die Erkrankung zu therapieren [103] [104].
Eine engmaschige Betreuung durch Pulmologen ist erforderlich. Regelmäßige Spirometrien, Sputum-Kulturen und Röntgen-Thorax Kontrollen werden empfohlen. Vor allem bei Kindern ist die regelmäßige Hals-Nasen-Ohrenärztliche Kontrolle aufgrund der rezidivierenden Otitiden und daraus resultierenden Schallleitungsstörungen erforderlich.
Nasale Symptome manifestieren sich in der Regel als Rhinorrhoe und Nasenatmungsbehinderung. Eine Polypenbildung bei betroffenen Kindern ist selten. Prophylaktische Antibiotika helfen, die infektiöse Komponente der Rhinosinusitis zu reduzieren. Die Indikation zur Nasennebenhöhlenoperation sollte zurückhaltend gestellt werden, da deren Wirksamkeit umstritten ist. Es gibt keine Evidenz für den Nutzen intranasaler Steroide, allerdings können sie bei der Behandlung einer zusätzlichen allergischen Rhinosinusitis hilfreich sein. Verbesserte genetische Diagnosestellungen sind der erste Schritt hin zu einer zukünftigen, genbasierten Therapiestrategie wie z. B. Genersatztherapie, Aminoglykosid-induziertes „translationales Überlesen“ (read-through) und pharmakogenetischen Ansätzen [105].
2.4 Tumore
Tumore des Sinunasaltrakts und der vorderen Schädelbasis können primär in dieser Region entstehen oder ihren Ursprung an einer entfernteren Stelle der Kopf-Hals-Region, allerdings eine Manifestation innerhalb des Sinunasaltrakts oder an der Schädelbasis aufweisen. In der Einteilung der World Health Organization von 2017 wird auf eine weitere Gruppe von Neoplasien verwiesen, deren Auftreten innerhalb des Sinunasaltrakts und der vorderen Schädelbasis aus differenzialdiagnostischen Gründen von Bedeutung ist. Die folgende Einteilung benigner und maligner Entitäten des Sinunasaltrakts und der vorderen Schädelbasis entstand in Anlehnung an diese Klassifikation der WHO [106].
2.4.1 Benigne Tumore
Benigne Tumore des Sinunasaltrakts werden in drei Hauptkategorien eingeteilt, die im folgenden Teil dargestellt werden:
-
Weichteil-, Nerven- und Gefäßtumore
-
Ossäre Tumore
-
Andere Weichteiltumore
2.4.1.1 Weichteil-, Nerven- und Gefäßtumore
2.4.1.1.1 Schleimhautpapillome
Drei unterschiedliche Varianten von Schleimhautpapillomen werden unterschieden. Allen gemeinsam ist die Entstehung aus der sogenannten Schneider-Membran, die Nasenhaupt- und Nasennebenhöhlen auskleidet [107] [108] [109] [110]. Diese ektodermal entstandene, mit Zilien besetzte Mukosa entwickelt sich als eine Invagination von olfaktorischem Ektoderm in der vierten Woche der embryonalen Entwicklung [111]. Aus pathologischer Sicht werden 3 Schneider-Papillome unterschieden:
-
Invertiertes Papillom
-
Exophytisches Papillom
-
Onkozytäres Papillom
Exophytisches Papillom:
Diese Entität ist auch als fungiformes oder septales Papillom bekannt und stellt 6–50% aller Schneider-Papillome. Im Gegensatz zum invertierten Papillom treten sie vorwiegend bei Männern im Alter zwischen 20 und 50 Jahren auf. Manifestationsort ist in den meisten Fällen das anteriore Nasenseptum, gelegentlich auch die laterale Nasenwand. Multifokales Auftreten ist möglich, bilaterale Manifestationen wurden sehr selten beschrieben [112]. Eine Manifestation in den Nasennebenhöhlen ist äußerst selten. Makroskopisch imponiert das exophytische Papillom als rosig bis graue Raumforderung mit gefältelter Oberfläche. Die Therapie der Wahl ist die Exzision. Eine maligne Entartung wurde nicht beschrieben.
Onkozytäres Papillom:
Diese Entität ist mit 2–26% die seltenste Ausprägung der Schneider-Papillome. Die Geschlechterverteilung ist annähernd gleich, eine Manifestation ist meist nach der fünften Dekade feststellbar. Die onkozytären Papillome entstehen ausschließlich an der lateralen Nasenwand, dem Sinus ethmoidalis oder maxillaris [113] [114]. Sie sind dem invertierten Papillom in vielen Punkten sehr ähnlich, sodass einige Autoren die onkozytären Papillome als mikroskopische Variante des invertierten Papilloms bezeichnen [111] [115] [116]. Eine maligne Entartung ist – analog zum invertierten Papillom – möglich (siehe folgendes Kapitel).
Invertiertes Papillom:
Invertierte Papillome repräsentieren mit 47–78% die häufigste Entität der Schneider-Papillome. Sie zeigen ein polypöses, meist gelapptes Wachstum, mikroskopisch in das Stroma der Mukosa eingestülpte Epithelzapfen. Entstehungsort ist in 48% der Sinus ethmoidalis, in 28% der Sinus maxillaris, in 7,5% der Sinus sphenoidalis und in 2,5% der Sinus frontalis. Auch Manifestationen an der Schleimhaut des Nasenseptums sind möglich. Typischerweise ist eine unilaterale Manifestation festzustellen. Eine bilaterale Manifestation ist äußerst selten beschrieben [117] [118]. Eine sekundäre metachrone Malignisierung ist in bis zu 4% der invertierten Papillome dargestellt mit dem Plattenepithelkarzinom als häufigste Entität, die bei Rezidiven eines invertierten Papilloms auf bis zu 11% ansteigt [119] [120] [121].
Die Inzidenz des invertierten Papilloms liegt zwischen 0,5 und 1,5:100 000 Einwohnern pro Jahr mit einem Altersgipfel zwischen der 5. Und 6. Dekade. Männer sind häufiger betroffen (M:W 2–5:1) [108] [118] [122].
Patienten mit invertiertem Papillom berichten von Nasenatmungsbehinderung, Epistaxis und Epiphora, sofern eine Affektion der ableitenden Tränenwege oder des unteren Nasengangs vorliegt. Je nach Lokalisation und invasivem Wachstumsverhalten kann es zu der Ausbildung einer Mukozele oder einer Protrusio bulbi kommen.
Klinisch präsentiert sich meist eine ödematöse, eher durchsichtig wirkende Polypenformation. Allerdings unterliegt das Erscheinungsbild starken Variationen, da die polypöse Raumforderung auch entzündlich und fleischig imponieren kann [123].
Die Computertomografie ist die bildgebende Modalität der Wahl, da knöcherne Erosionen, die auf eine Maligne Transformation hinweisen, dargestellt werden. Häufig ist am Entstehungsort des invertierten Papilloms eine Hyperostose oder Sklerose der knöchernen Grenzen zu erkennen. Auch kann eine Kalzifikation innerhalb der Raumforderung vorkommen [124].
Die Exzision mit der unmittelbar umgebenden Schleimhaut ist Therapie der Wahl. Ein Sicherheitsabstand von 1–1,5 cm sollte berücksichtigt werden [62] [125] [126]. Zudem ist die Abtragung bzw. Ausdünnung des Knochens im Bereich der Ansatzstelle erforderlich. Rezidive treten bei unzureichender Resektion deutlich häufiger auf [127]. Bei defensiver Polypektomie oder lokaler Exzision wurden in bis zu 78% der Fälle Rezidive beschrieben [128]. Innerhalb der letzten Jahre haben sich durch die mediale Maxillektomie und ihre Variationen effizientere Zugangswege zu allen Bereichen der Kieferhöhle etabliert [64]. Diese Technik hat den ursprünglichen Caldwell-Luc Zugang in den meisten Fällen ersetzt [129] [130]. Eine Übersichtsarbeit mit Positionspapier zeigte gleichwertige, wenn nicht sogar bessere Ergebnisse für die endoskopische Resektion von invertierten Papillomen. Bei der Lokalisation in der Kieferhöhle ist der endonasale Zugang dem offenen Vorgehen hinsichtlich Rezidiv-Häufigkeit überlegen [121] [131]. Bei Verwendung eines prälacrimalen Zugangs ist die Rezidivrate noch geringer [125]. Grundsätzlich sind die langfristigen Ergebnisse umso besser, je gründlicher die erkrankte Schleimhaut reseziert wird [118].
2.4.1.1.2 Leiomyome
Leiomyome sind gutartige Tumore mit muskulärer Differenzierung. Bei Angioleiomyomen besteht zudem eine vaskuläre Differenzierung.
In der Kopf-Hals-Region sind sie extrem selten und repräsentieren insgesamt nicht einmal 1% aller insgesamt vorkommenden Leiomyome [132]. In den meisten Fällen sind die Patienten Erwachsene ohne Bevorzugung eines Geschlechts. Die meisten sinunasal vorkommenden Leiomyome weisen eine vaskuläre Differenzierung auf [106] [132] [133].
Das Tumorwachstum ist lange unbemerkt und sorgt zumeist für unspezifische Symptome wie Druckgefühl und langsam progredienter Nasenatmungsbehinderung. Epistaxis und Schmerzen können hinzukommen.
Makroskopisch präsentieren sich nasale Leiomyome polypoid bis nodulär und gut abgrenzbar mit einer weißlich/bräunlichen Schnittfläche. Die Raumforderung liegt meist unter intakter Schleimhaut. Ulcerationen treten selten auf. Spindelförmige Tumorzellen sind in sich überlappenden Faszikeln angeordnet mit ovalen, länglichen, zigarrenförmig anmutenden Zellkernen ohne Atypien. Eosinophiles, fibrilläres Zytoplasma ist vorhanden. Im Gegensatz zum Leiomyosarkom existieren keine mitotischen Veränderungen. Das Angioleiomyom besitzt zusätzlich prominente Gefäße, die von Muskelzellen umgeben und eng mit ihnen verbunden sind [106].
Trotz des seltenen Vorkommens existiert die Möglichkeit einer malignen Transformation hin zu einem Leiomyosarkom. Insofern sollte eine Verzögerung der Therapie vermieden werden. Therapie der Wahl ist die Tumorresektion. Die Prognose ist bei vollständiger Exzision äußerst gut. Rezidive sind in diesem Fall extrem selten [134] [135] [136].
2.4.1.1.3 Hämangiome
Das lobuläre kapilläre Hämangiom wurde durch die Franzosen Poncet und Dor 1897 erstmals beschrieben und als Botryomycosis hominis bezeichnet [137]. Ursprünglich wurde vermutet, die Erkrankung sei von Pferden auf Menschen übertragen, was allerdings durch Hartzell einige Jahre später widerlegt wurde [138]. Heutige Synonyme sind Granuloma pyogenicum, kapilläres Hämangiom und Epulis gravidarum.
Mukosale Hämangiome repräsentieren ca. 10% aller Kopf-/Hals-Hämangiome und ca. 25% aller nichtepithelialen Neoplasien des Sinunasaltrakts [106] [139] [140].
Ihren Ursprung nehmen die Hämangiome in Kapillaren, wenn deren Dichte zu groß wird und diese trotzdem ihre ursprüngliche Architektur mit Stamm und Verästelungen sowie umgebenden Perizyten behalten. Aufgrund der Assoziation mit Traumata bzw. Manipulationen und hormonellen Umstellungen im Rahmen von Schwangerschaften wird eine reaktive Entstehung der lobulären kapillären Hämangiome diskutiert [141]. Ein gehäuftes Auftreten wird zudem bei Anwendung des Proteinkinase-Inhibitors Vemurafenib berichtet [142].
Das lobuläre kapilläre Hämangiom (Granuloma pyogenicum) tritt in allen Altersstufen auf, zeigt allerdings eine Häufung bei Kindern und adoleszenten Männern sowie Frauen im gebärfähigen Alter. Jenseits der 4. Dekade ist die Geschlechterverteilung gleich.
Die Läsionen können eine Größe von bis zu 5 cm erreichen. Die Oberfläche ist rotbläulich unter intakter Mukosa gelegen. Der Tumor tastet sich weich, ist kompressibel und mutet gelegentlich polypös an.
Histologisch zeigen lobuläre kapilläre Hämangiome ein stamm- und astartig verzweigtes Muster aus kapillärer Proliferation, das von Perizyten umgeben ist. Die einzelnen Lobuli sind durch ein fibromyxoides Stroma voneinander getrennt. Inflammatorische Infiltrate treten vor allem bei ulzerierten Oberflächen auf [141].
Primäres klinisches Symptom ist die unilaterale Epistaxis gefolgt von einer schmerzlosen, obstruktiven Raumforderung. Häufigste Manifestationsorte sind vorderes Nasenseptum und der Kopf der unteren Nasenmuschel. Ein Auftreten in den Nasennebenhöhlen ist ebenfalls möglich ebenso wie der Befall der äußeren Nase.
Therapie der Wahl ist die Tumorexzision. Ausgedehntere Befunde können einer präoperativen Embolisation zugeführt werden, um das Blutungsrisiko zu verringern. Bei schwangeren Patientinnen ist eine Regression nach Geburt zu erwarten. Multiple Rezidive sind bei unvollständiger Resektion vor allem bei Kindern zu erwarten [143].
2.4.1.1.4 Schwannome
Schwannome sind gutartige Tumore, die von Schwann-Zellen ausgehen. Synonym werden die Begriffe Neurilemmom und benigner peripherer Nervenscheidentumor verwendet.
25–45% aller Schwannome entstehen in der Kopf-Hals-Region. Am häufigsten in genanntem Bereich finden sich Manifestationen entlang des N. vestibulocochlearis. Nur etwa 4% aller Schwannome manifestieren sich im Sinunasaltrakt. Laut Orpha.net liegt die Inzidenz von benignen Schwannomen bei 6:100 000, die sinunasale Ausprägung ist damit als noch wesentlich geringer anzunehmen. Die Altersspanne der sinunasalen Manifestationen ist mit 17–81 Jahren weit gefasst und zeigt einen Altersgipfel bei 50 Jahren auf ohne Bevorzugung eines Geschlechts [106] [144].
Ursprünge sind entlang der Äste der Hirnnerven V und IX sowie des autonomen Nervensystems zu finden. Die entstehenden Tumore können die Nasenhaupthöhle und alle Nasennebenhöhlen betreffen [144] [145].
Primäre Symptome sind Nasenatmungsbehinderung, Epistaxis, Hyposmie und gelegentlich Schmerzempfindungen sowie die Ausprägung eines Horner Syndroms. Bildgebende Verfahren zeigen eine inhomogene Tumormasse niedriger Dichte mit gelegentlicher Knochenarrosion. Differentialdiagnostische Überlegungen schließen daher Ästhesioneuroblastome, adenoidzystische Karzinome und Plattenepithel-Karzinome mit ein. ([Abb. 9]) zeigt eine computertomografische Darstellung eines großen Schwannoms des Nervus maxillaris, welches den Ursprung im Foramen rotundum hat. Magnetresonanztomografisch zeigt sich ein in der T1-Wichtung hyperintenses, inhomogenes Binnensignal der Raumforderung.

Schwannome haben eine kugelförmige, gut abgrenzbare Konfiguration. Der Tumor tastet sich fest und zeigt eine gelblich-bräunliche, teils zystischen Oberfläche [106]. Histologisch sind Schwannome unbekapselte Tumore, die sich aus zellreichen, sogenannten Antoni A Arealen mit nukleären Palisaden sowie zellarmen, myxoiden Antoni B Arealen zusammensetzen. Die Tumorzellen zeigen ein fusiformes Bild mit zystoplasmatischen Ausziehungen, das einen wellige bis spindelige Erscheinung verleiht. Mitosen sind äußerst spärlich, Nekroseareale existieren nicht [106].
In Ausnahmefällen ist eine Entartung eines Schwannoms möglich. Nach vollständiger Tumorresektion sind Rezidive sehr selten. Aufgrund des sehr langsamen Tumorwachstums ist eine subtotale Tumorresektion in Fällen von starker Adhärenz zu kritischen neurovaskulären Strukturen möglich [144]. Innerhalb des Tumors verlaufende Fasern des betroffenen Nerven sind in der Regel funktionslos, weshalb eine Resektion zu keinen neurologischen Ausfällen führt [146]. In Fällen von zervikalen Schwannomen wird die intrakapsuläre Dissektion empfohlen, um die Nervenfaszikel, welche den Tumor umgeben, nicht zu zerstören [147]. Aufgrund der beengten Platzverhältnisse und der engen Zugangswege ist dies bei Schwannomen der Nasennebenhöhlen allerdings nur sehr eingeschränkt möglich.
2.4.1.1.5 Neurofibrome
Neurofibrome sind gutartige periphere Nervenscheidentumore aus Schwann-Zellen, perineuralartigen Zellen und intraneuralen Fibroblasten. Synonym wird der Begriff Fibroneurom verwendet.
Sinunasale Manifestationen von Neurofibromen sind äußerst selten und können in allen Altersklassen auftreten. Der Altersgipfel betroffener Patienten liegt bei 50 Jahren. Für Patienten mit einer Neurofibromatose Typ I liegt er bei 35 Jahren [148]. Die Prävalenz der Neurofibromatose liegt bei 21:100 000. Letztere sind für ca. 10% der sinunasalen Neurofibrome verantwortlich.
Häufigste Lokalisationen sind Naseneingang und Sinus maxillaris bei überwiegend unilateraler Manifestation. Beschwerden betroffener Patienten sind Nasenatmungsbehinderung, Epistaxis und Schmerzen über den betroffenen Arealen [148] [149].
Neurofibrome zeigen eine glänzende, fusiforme und manchmal polypoide Oberfläche und sind fest palpabel [148] [149].
Auch Neurofibrome sind unbekapselte Tumore, die eng mit Nervenästen assoziiert sind. Modifizierte Schwannzellen, intraneurale Fibroblasten und perineurale Hybridzellen mit gröberen Kollagensträngen sowie Mastzellen in einem mukopolysaccharid-reichen Stroma bestimmen das histopathologische Bild. Ovaläre bis spindelförmige Zellen mit undulierenden, spitz zulaufenden Zellkernen mit dünnen zytoplasmatischen Prozessen, die sich in das Stroma ausdehnen, sind vorhanden.
Die Prognose ist bei vollständiger Tumorresektion sehr gut. Rezidive treten in 5% v. a. bei inkompletten Resektionen auf. Eine Malignisierung ist äußerst selten [150].
2.4.1.1.6 Meningeome
Meningeome sind gutartige Tumore meningothelialen Ursprungs. Sinunasale Meningeome gehen von extrakraniellen, versprengten arachnoiden Zellen innerhalb von Nervenscheiden oder Gefäßen aus.
Sinunasale Meningeome sind äußerst selten und für weniger als 0,1% aller primären sinunasalen Neoplasien, 2% aller Meningeome und 24% aller extrakraniellen Meningeome verantwortlich. Differenziert werden müssen sie von intrakranialen Befunden mit extrakranieller Ausbreitung in den Sinunasal-Trakt.
Patienten jeden Alters sind betroffen und es existiert – entgegen den intrakraniellen Meningeomen – keine Prädilektion für das weibliche Geschlecht. Das mittlere Erkrankungsalter beträgt 48 Jahre bei einer Altersspanne von 13–88 Jahren.
Manifestationen finden sich häufig in der Nasenhaupthöhle als auch den Nasennebenhöhlen. Eine Manifestation in nur einem der beiden Orte ist noch seltener. Interessanterweise sind die meisten derartigen Tumore linksseitig lokalisiert [151] [152] [153].
Die Patienten fallen häufig mit einer endonasalen, polypös anmutenden Raumforderung, mit Nasenatmungsbehinderung, Epistaxis, sinunasalen Beschwerden, Schmerzen, Cephalgien, Exophthalmus, periorbitalen Ödemen oder Visusstörungen auf [106].
Knocheninfiltrationen und Schleimhautulzerationen sind möglich. Die Schnittfläche des Tumors zeigt eine grau-weißliche, bräunliche oder rötliche Farbe. Kalzifikationen und Knochenfragmente können häufig identifiziert werden.
Mikroskopisch können extrakranielle Meningeome äußerst unterschiedliche Erscheinungsbilder aufweisen. Häufig zeigen sie meningotheliomatöses Wachstum mit undeutlichen Zellgrenzen. Intranukleäre Pseudoeinschlüsse und Psammomkörper sind häufig.
Von den 15 beschriebenen histologischen Typen von Meningeomen kommen im sinunasal-Trakt meningotheliale, transitionale, metaplastische und psammomatöse Tumore vor. Die meisten Tumore können dem WHO Grad I zugeteilt werden. Sinunasale Manifestationen von Meningeomen Grad II (atypische Meningeome mit schnellem Wachstum) oder Grad III (anaplastische Meningeome, infiltratives Wachstum) sind extrem selten [154] [155].
Differenzialdiagnosen schließen Karzinome, Melanome, aggressive psammomatöse ossifizierende Fibrome ein.
Die vollständige chirurgische Exzision ist Therapie der Wahl, auch wenn Rezidivraten von bis zu 30% berichtet werden. Bei Meningeomen, die nicht vollständig resektabel sind, kann eine Radiotherapie zur Wachstumshemmung führen [156] [157]. Die insgesamte Prognose sinunasaler Meningeome ist gut. Metastasierungen und maligne Transformationen werden nicht beschrieben [106].
2.4.1.2 Ossäre Tumore
2.4.1.2.1 Osteome und Gardner Syndrom
Osteome sind gutartige, langsam wachsende Tumore des Schädelknochens, die sich häufig in den Nasennebenhöhlen und der Schädelbasis manifestieren. Am häufigsten finden sich die Knochenwucherungen im Sinus frontalis (70–80%), dem Sinus ethmoidalis (20–25%), dem Sinus maxillaris (5%) und extrem selten dem Sinus sphenoidalis. Die Inzidenz von Osteomen in den Nasennebenhöhlen liegt bei ca. 3% [118] [158] [159]. Damit sind Osteome per definitionem keine seltene Erkrankung der Nasennebenhöhlen. Sie können allerdings ein Symptom im Rahmen des Gardner Syndroms sein, weshalb sie in der vorliegenden Arbeit trotzdem thematisiert werden.
Die Ätiologie der Tumore ist nicht gänzlich geklärt. Aktuellen Theorien vermuten embryologische Fehlbildungen, traumatische oder entzündliche Trigger, genetische Prädisposition und Störungen des Calcium-Stoffwechsels als Ursache [118] [158] [160].
Nur ca. 10% aller Osteome der Nasennebenhöhlen werden symptomatisch. Beschwerden von betroffenen Patienten sind häufig assoziiert mit Obstruktionen der Drainagewege der Nebenhöhlen, also rezidivierenden akuten Sinusitiden und auch chronisch sinunasalen Beschwerden. Druckgefühl, Gesichtsschmerzen und Rhinorrhoe sind klassische Symptome. Bei Beteiligung benachbarter Strukturen der Nasennebenhöhlen kann es zu Kompressionen der Orbita oder des Nervus opticus sowie bei intrakranieller Beteiligung zu einem Pneumatocephalon kommen [158] [161].
Die Computertomografie zeigt hyperdense, homogene, gut abgrenzbare Areale. Die Magnetresonanztomografie kann helfen, ossifizierende Fibrome oder eine fibröse Dysplasie differentialdiagnostisch auszuschließen [118].
Asymptomatische Osteome sollten entsprechend aktuellem Konsens mittels „wait and scan“ behandelt werden [162] [163] [164]. Regelmäßige computertomografische Kontrollen alle 2 Jahre geben Aufschluss über Wachstumsgeschwindigkeit des Tumors [165]. Bei symptomatischen Osteomen ist die möglichst vollständige Resektion Methode der Wahl. Je nach Lokalisation kommen endonasale oder offene chirurgische Verfahren zum Einsatz.
Gardner-Syndrom: Das Gardner-Syndrom zählt mit einer Inzidenz von 1:8 000 zu den seltenen Erkrankungen [166]. Die Prävalenz in den USA liegt derzeit bei ca. 1:1 000 000 Einwohnern. Patienten, die vom Gardner Syndrom betroffen sind, zeigen häufig (multiple) Osteome, Weichteiltumore und eine intestinale Polyposis (v. a. im Colon) auf. Das Gardner Syndrom wird autosomal dominant vererbt.
Ein genetischer Zusammenhang mit der Entwicklung des Gardner-Syndroms wurde in einer Mutation des Gens für adenomatöse Polyposis coli (APC) gezeigt, das auf Chromosom 5 liegt. Dieses Tumorsuppressorgen ist für die Produktion des APC-Proteins verantwortlich, welches das Zellwachstum im Zellzyklus reguliert [167] [168] [169].
Regelmäßige Coloskopien sind bei betroffenen Patienten obligat. Bei Nachweis der APC-Genmutation gilt die Entwicklung eines Colonkarzinoms ab einem Alter von 40 Jahren als sicher, weshalb bei Vorliegen von 20 oder mehr Colon-Polypen eine Colektomie empfohlen wird [166]. Insofern sollte bei Vorliegen multipler Osteome differenzialdiagnostisch immer an das Vorliegen des Gardner Syndroms gedacht werden.
2.4.1.2.2 Fibröse Dysplasie
Bei der Fibrösen Dysplasie besteht eine mesenchymale Knochenentwicklungsstörung, die durch postzygotisch somatisch aktivierende Mutationen verursacht wird. Es kommt zu einer Aktivierung der Adenylylzyklase und Erhöhung von zyklischem AMP, das auf die nachgeschalteten Signalwege wirkt und den Ersatz von normalem Knochen durch fibröses Gewebe und abnormal strukturierten Knochen bewirkt [170]. Die Inzidenz wird mit 1:4 000–10 000 angegeben [171] [172].
Je nach Lokalisation der Knochenherde berichten die Patienten von Cephalgien und Druckgefühl. Bei Lokalisation an der Tabula externa des Schädelknochens kommt es zu sichtbaren Deformitäten und entsprechend kosmetischer Beeinträchtigung. Manifestationen an den Ostien oder Schlüsselstellen der Nebenhöhlen-Drainagewege können zu sinunasalen Beschwerden führen, die die zugrundeliegende Erkrankung lange maskieren.
Aufgrund der Seltenheit der Erkrankung existieren nur wenig Daten zur Therapie der fibrösen Dysplasie. Eine kausale Therapie existiert bislang nicht. Es wurde über Schmerzreduktion durch Gabe von Bisphosphonaten berichtet [173] [174] [175]. Eine Supprimierung der Osteoblastenaktivität konnte allerdings nicht nachgewiesen werden [176]. Bei asymptomatischem Verlauf wird die primär observierende Therapie favorisiert. Funktionseinschränkungen und Beschwerden wie Cephalgien rechtfertigen ein operatives Vorgehen in Abhängigkeit der Lokalisation der Manifestation. Eine prophylaktische operative Therapie wird nicht empfohlen, stattdessen sollten entsprechend der verfügbaren Daten regelmäßige bildmorphologische Verlaufskontrollen erfolgen [177] [178].
2.4.1.2.3 McCune-Albright-Syndrom
Das McCune Albright Syndrom wurde zuerst 1936 als eine Trias von fibröser Dysplasie, Café-au-Lait-Flecken der Haut und Pubertas praecox beschrieben [179]. Der heute bekannte, deutlich variablere Phänotyp macht das McCune Albright Syndrom zu einer interdisziplinäreren Herausforderung.
Ursächlich für die Ausbildung des Syndroms ist eine Mutation des GNAS1-Gens (Guanine nucleotide binding protein alpha stimulating activity polypeptide 1), welches auf Chromosom 20 lokalisiert ist.
Die Seltenheit dieser Erkrankung führt zu einer nur unzureichenden Charakterisierung der sinunasalen Beteiligung von Patienten mit McCune-Albright-Syndrom. Derzeit bekannte Symptome stammen vorwiegend aus Fallberichten, die Komplikationen der Erkrankung schildern. Die Prävalenz der Erkrankung wird auf 0,55:100.000 geschätzt [2].
Patienten mit McCune-Albright-Syndrom schildern primär Schwäche in den Extremitäten oder Schmerzempfindungen. Die am häufigsten betroffene Lokalisation ist der proximale Femur. Frakturen der betroffenen Knochenareale finden häufig in der Kindheit statt mit sinkender Inzidenz in Richtung Adoleszenz [180]. Charakteristisch sind die Deformierungen des Knochens unter Belastung, die zu den für die Erkrankungen typischen kraniofazialen Stigmata führen. Kraniofaziale Ausbildungen der fibrösen Dysplasie weisen ein langsames Wachstum mit schmerzloser Schwellung auf, die zu einer bisweilen deutlichen Asymmetrie des Mittelgesichts führt ([Abb. 10] und [11]). Milde Verläufe werde häufig zufällig im Rahmen von Röntgenaufnahmen der Zähne und im Rahmen von bei Polytraumen durchgeführten Computertomografien diagnostiziert [170]. Schwerere Verläufe führen häufiger zu Schmerzempfindungen, Parästhesien, Okklusionsstörungen, Hörbeeinträchtigungen und Visusstörungen [170] [178] [181] [182]. Betroffene Patienten zeigen in bis zu 50% Beeinträchtigungen der Schilddrüsenfunktion, meist eine Hyperthyreose. GNAS führt in ca. 15% der Patienten zu einer Steigerung von Prolaktin und GH in der Hypophyse. Letzteres führt zu den charakteristischen kraniofazialen Veränderungen, die bei den meisten Patienten ersichtlich sind [183].


DeKlotz et al. konnten in einem Kollektiv von 130 Patienten mit McCune-Albright Syndrom bei 112 Patienten kraniofaziale Veränderungen nachweisen [182]. 33% berichteten von Cephalgien oder Gesichtsschmerzen, Nasenatmung war in 29% der Fälle und chronisch sinunasale Beschwerden sowie Hyposmie in jeweils 7% festzustellen. Ein Progress der sinunasalen Beteiligung der Fibrösen Dysplasie nach Abschluss der Adoleszenz ist selten. Schwerwiegende Komplikationen im Rahmen des normalen Fortschreitens der Erkrankung sind ebenfalls selten anzutreffen ([Abb. 12]).
Wie bei der Fibrösen Dysplasie existiert keine kausale Therapie. Bisphosphonate zeigten keinen relevanten Therapieerfolg [184] [185] [186]. Gemäß allgemeinem Konsens wird das konservative Vorgehen häufiger favorisiert, wohingegen chirurgische Maßnahmen bei signifikanten Symptomen oder bei Kompression vitaler Strukturen zum Einsatz kommen [187] [188] [189] [190].

2.4.1.3 Andere Weichteiltumore
In der folgenden Sektion werden seltene benigne Entitäten des Sinunasaltrakts vorgestellt, die epithelialen, odontogenen, neuroglialen Ursprungs sind.
-
Respiratorisches epitheliales adenomatoides Hamartom
-
Sinunasales Ameloblastom
-
Chondromesenchymales Hamartom
-
Nasales Gliom
-
Cholesteatom
2.4.1.3.1 Respiratorisches epitheliales adenomatoides Hamartom (REAH)
Das respiratorische epitheliale adenomatoide Hamartom wurde erstmals 1995 beschrieben [191]. Im Rahmen dieser Erkrankung kommt es zu einer tumorartigen Proliferation von Drüsen im Stroma, welches von mehrreihigem Flimmerepithel bedeckt ist. Manifestationen finden sich in den Nasennebenhöhlen, der Nasenhaupthöhle und dem Nasenrachen. Uni- oder bilaterale Manifestationen sowie Assoziation mit chronisch polypöser Rhinosinusitis sind möglich. Die Zahl der weltweit beschriebenen Fälle schwankt zwischen 60 [191] [192] [193] [194] [195] [196], bis ca. 200 [197] [198]. Das Syndrom zählt aufgrund der niedrigen Inzidenz zu den seltenen Erkrankungen.
Vor allem Männer zwischen 30 und 90 Jahren sind von der Neubildung betroffen [191] [195]. Die Symptome entsprechen üblicherweise denen einer chronischen Rhinosinusitis, also Druckgefühl und Nasenatmungsbehinderung, Epistaxis und Rhinorrhoe, Gesichtsschmerzen und Hyposmie [191] [192] [193] [199] [200].
Am häufigsten manifestiert sich das epitheliale adenomatoide Hamartom in der Nasenhaupthöhle, vor allem am posterioren Nasenseptum. Nicht selten kommt es zu einem Befall beider Seiten [191] [192] [193] [194] [195] [196] [199]. Häufig wird es im Rahmen einer Polyposis nasi diagnostiziert. Bei Manifestation an der Fossa olfactoria kann es zu einer deutlichen Erweiterung derselben kommen, was differenzialdiagnostisch hinsichtlich einer Polyposis nasi relevant ist [201] [202]. Eine Erweiterung der Fossa olfactoria bei einem lokalen Befall, fehlende Kontrastmittelanreicherung in der Computertomografie und ansonsten bildmorphologisch unauffällige Nasennebenhöhlen sollten daher unbedingt an ein epitheliales adenomatoides Hamartom denken lassen.
Aufgrund des primären Eindrucks wird die Diagnose häufig im Rahmen einer Biopsie gesichert. Therapeutisch ist die vollständige Exzision anzustreben. Rezidive sind in den verfügbaren Studien innerhalb eines Zeitraums von 5 Jahren nicht beobachtet worden [191] [193] [199] [203] [204].
2.4.1.3.2 Sinunasales Ameloblastom
Ameloblastome sind gutartige, aber aggressive odontogene Tumore, die in den meisten Fällen den Unterkiefer betreffen. Nur etwa 15% treten am Oberkiefer auf. Als eine Untergruppe sind die sinunasalen Ameloblastome zu betrachten, die vermutlich von der epithelialen Auskleidung der Nasennebenhöhlen abstammen. Manifestationsorte sind die Nasennebenhöhlen, in einigen Fällen auch mit Beteiligung der Nasenhaupthöhle.
Es zeigt sich eine meist schmerzlose Raumforderung, die im weiteren Verlauf zu Nasenatmungsbehinderung und Druckgefühl führen kann. Im Unterschied zu Ameloblastomen des Kiefers zeigt die Sinunasale Variante radiografisch ein solides Bild mit teilweisen Verschattungen [205].
Histologisch gesehen sind sinonasale Ameloblastome identisch mit Manifestationen der Mundhöhle, mit klassischen Merkmalen palisadierter säulenförmiger Basalzellen, die eine zentrale Proliferation umgeben, die dem sternförmigen Retikulum eines sich entwickelnden Zahnes ähnelt. In den Nasennebenhöhlen kann eine ameloblastomartige Proliferation unter intakter Mukosa dargestellt werden. Dieser Befund ist – bei gleichzeitig nicht vorhandener Verbindung zum Kiefer – eine Bestätigung für einen primär sinunasalen Ursprung des Tumors. Im Gegensatz zu den gnathischen Variante ist die Prävalenz des sinonasalen Ameloblastoms bei Männern höher. Die Inzidenz wird insgesamt auf ca. 0,5:100 000 000 geschätzt [206] [207]. Der Altersgipfel liegt bei ca. 60 Jahren [208] und damit etwa 15–20 Jahr höher als bei der am Kiefer lokalisierten Variante [106].
Der Behandlungserfolg und damit die Rezidivfreiheit ist abhängig von der vollständigen chirurgischen Exzision des Befundes. Detaillierte bildgebende Darstellungen sind essentiell, um Residuen zu vermeiden. Rezidive treten in den meisten Fällen innerhalb von 1–2 Jahren auf, können allerdings auch nach deutlich längeren Zeiträumen entstehen [205]. Beschreibungen von Mortalität durch sinunasale Ameloblastome, Metastasierungen oder maligne Transformationen existieren in der aktuellen Literatur nicht.
2.4.1.3.3 Chondromesenchymales Hamartom
Das chondromesenchymale Hamartom ist eine gutartige, langsam wachsende Raumforderung mit lokal destruktivem, tumorähnlichem Wachstum und unterschiedlichen mesenchymalen Anteilen. Synonym werden die Begriffe nasales chondromesenchymales Hamartom und Mesenchymom verwendet.
Der Tumor ist sehr selten und tritt meist bei Kindern auf, nur selten auch bei älteren Kindern und Erwachsenen mit einer leichten Bevorzugung des männlichen Geschlechts. Es existieren in der derzeitigen Literatur ca. 60 publizierte Fälle [209].
Nasennebenhöhlen, Nasenhaupthöhle und Orbita können betroffen sein. Ausdehnungen entlang der Schädelbasis und nach intrakraniell sind möglich [209] [210].
Symptome betroffener Patienten sind Nasenatmungsbehinderung und Druckgefühl über der betroffenen Region. In der Bildgebung kann aufgrund des destruktiven Wachstums mit Knochenarrosionen der Eindruck eines malignen Geschehens entstehen.
Makroskopisch erinnert das feste und weißliche Gewebe an Knorpel. Mikroskopisch zeigt sich ein lobuläres Proliferationsmuster aus reifem und unreifem hyalinem Knorpel mit unterschiedlichem zellulärem und fibrösem Hintergrund. Die Knorpel- und Stroma-Anteile können mit knöchernen trabekulären Strukturen durchsetzt sein, oder knöcherne Inseln umgeben [106] [211].
Nach vollständiger Tumorresektion ist die Rezidivrate sehr gering und die Prognose entsprechend gut.
2.4.1.3.4 Nasales Gliom
Nasale Gliome (Synonym: heterotopes, ZNS-Gewebe) sind Ansammlungen von heterotopem neuroglialen Gewebe. Manifestationen können in der Nase und an der äußeren Nase auftreten. In 60% zeigt sich die Raumforderung am Nasenrücken, in 30% innerhalb der Nasenhaupthöhle. Noch seltener, in ca. 10% der Fälle, werden Raumforderungen an beiden genannten Stellen diagnostiziert, die durch einen Defekt im Knochen miteinander in Verbindung stehen [212].
Bei den meisten Patienten besteht die Raumforderung bereits bei der Geburt. Ca. 90% der Fälle werden im Alter von ca. 2 Jahren diagnostiziert bei einer gleichmäßigen Verteilung über beide Geschlechter.
Klinisch zeigt sich eine gut abgrenzbare, glatte Raumforderung, die submukös lokalisiert und nicht kompressibel ist.
Bei innerhalb der Nasenhaupthöhle gelegenen Befunden kann es zu Nasenatmungsbehinderung kommen, die – neben der ästhetischen Beeinträchtigung eines am Nasenrücken gelegenen Befundes – Hauptsymptom des nasalen Glioms ist.
Weitaus seltenere Manifestationen finden sich in den Nasennebenhöhlen, Pharynx, Nasopharynx, Zunge, Gaumen, Tonsillen und innerhalb der Orbita [213].
Im Gegensatz zu paranasalen Zelen nehmen Gliome bei venöser Stauung nicht an Größe zu bzw. pulsieren nicht. Dies kann mit dem Fürstenberg-Test, bei dem eine Kompression der V. jugularis durchgeführt wird, differenziert werden. Zusätzlich zeigt sich in der Computer- oder Magnetresonanztomografie eine Weichgewebsmasse ohne intrakraniellen Anteil oder einen knöchernen Defekt am Übergang zur vorderen Schädelgrube.
Makroskopisch zeigt sich die Raumforderung als polypoide, weiche, graubräunliche Masse mit einer Größe von 1–3 cm. Mikroskopisch ist der Tumor unbekapselt und aus unterschiedlich großen Inseln von Gliagewebe zusammengesetzt. Dazwischen finden sich Astrozyten und bandartige Stränge vaskularisierten Bindegewebes. Das Gliagewebe geht fließend in das Stroma der Dermis über. Mitosen zeigen sich nicht.
Differenzialdiagnostisch müssen nasale Enzephalozelen ausgeschlossen werden, in denen sich im Vergleich zu nasalen Gliomen allerdings ZNS-Gewebe mit leicht aufzufindenden Neuronen befindet. Trotzdem kann es bei Rezidiven zu fibrösen Veränderungen des Tumors kommen, die eine genaue Differenzierung von einem nasalen Gliom stark erschweren.
Die vollständige Exzision des nasalen Glioms ist Therapie der Wahl. Bei inkompletten Resektionen kann es in bis zu 30% zu Rezidiven kommen. Lokal aggressives Verhalten oder eine Tendenz zur Entartung bestehen nicht [212].
2.4.1.3.5 Cholesteatom
Cholesteatome sind chronisch-eitrige Entzündungen durch versprengtes verhornendes Plattenepithel, welches zu Knochendestruktionen führt. Typischerweise findet sich diese Pathologie im Mittelohr, allerdings finden sich in der aktuellen Literatur auch knapp 30 Fallberichte mit Ausprägung innerhalb der Nasennebenhöhlen [214] [215]. Häufigster Manifestationsort ist die Stirnhöhle, gefolgt von Kieferhöhle und Siebbeinzellen. Ein aktueller Bericht präsentiert eine Manifestation in der Keilbeinhöhle [216]. In [Abb. 13] ist ein großes Cholesteatom der Keilbeinhöhle in einer magnetresonanztomographischen Darstellung zu erkennen. [Abb. 14] zeigt den intraoperativen, endoskopischen Befund.


Ursächlich können entweder kongenitale Zellversprengungen im Rahmen der Embryogenese oder sekundäre Versprengungen z. B. durch chirurgische Interventionen sein. Auch Metaplasien im Rahmen chronischer Entzündungen werden als Ursache diskutiert [215].
Die Beschwerden sind abhängig von der Lokalisation und können Cephalgien, Visusstörungen, Druckgefühl, Rhinorrhoe und Hiernnervenausfällen umfassen.
Therapie der Wahl ist die operative und möglichst vollständige Resektion, die aber je nach Lage nicht immer möglich ist. Ziel der Operation sollte zudem eine möglichst weite Drainagemöglichkeit der betroffenen Kavität sein, um postoperative Kontrollen und ggf. Reinigungen bei nicht vollständiger Resektion des Cholesteatoms zu ermöglichen.
2.4.2 Maligne Tumore
Malignome der Nasenhaupthöhle und der Nasennebenhöhlen repräsentieren 0,2–0,8% aller malignen Neoplasien des menschlichen Körpers [10]. Die Inzidenz von Malignomen in der Nasenhaupthöhle und den Nasennebenhöhlen wird mit unter 1,5:100 000 bei Männern und unter 1:100 000 bei Frauen angegeben. Folglich sind alle malignen Entitäten dieser anatomischen Region zu den seltenen Erkrankungen zu zählen.
2.4.2.1 Maligne epitheliale Tumore
2.4.2.1.1 Plattenepithelkarzinom
2.4.2.1.1.1 Verhornendes Plattenepithelkarzinom
Sinunasale Plattenepithelkarzinome sind maligne Neoplasien, die von der Oberfläche des Epithels der Nasenhaupthöhle und der Nasennebenhöhlen ausgehen und eine Plattenepithel-Differenzierung aufweisen.
Der Befall des Sinunasaltrakts durch Plattenepithelkarzinome ist selten. Er stellt die am wenigsten von dieser Entität betroffene Lokalisation der Kopf-Hals-Region dar [217]. Das Erkrankungsalter liegt zwischen dem 6. und 7. Lebensjahrzehnt mit deutlich mehr männlichen Patienten (m:w=2:1) [106] [217] [217] [219].
Tabakkonsum erhöht das Risiko des Auftretens eines Plattenepithelkarzinoms im Sinunasaltrakt, allerdings deutlich weniger als in anderen Lokalisationen der Kopf-Hals-Region [220] [220] [222]. High-risk HPV Infektionen sind meistens mit dem Auftreten des nichtverhornenden Plattenepithelkarzinoms assoziiert. In seltenen Fällen können sinunasale Papillome nach maligner Transformation in ein verhornendes oder in ein nicht verhornendes Plattenepithel übergehen [223].
Häufigster Manifestationsort ist der Sinus maxillaris, gefolgt von der Nasenhaupthöhle und dem Sinus ethmoidalis. Ein Befall des Sinus sphenoidalis oder frontalis ist äußerst selten [106].
Betroffene Patienten weisen initial unspezifische Beschwerden wie Nasenatmungsbehinderung, Epistaxis und Rhinorrhoe bzw. sinunasale Beschwerden auf. Schmerzen über der betroffenen Region, Protrusio bulbi oder Diplopie und Paralysen sind Symptome bei ausgedehnterem Befall. Bei älteren Patienten kann sich die Manifestation durch eine nicht mehr passende Oberkiefer-Zahnprothese bemerkbar machen, wenn der Hartgaumen infiltriert ist.
Makroskopisch wächst der Tumor exo- oder endophytisch mit variablen Ulzerationen, Nekrosearealen und hämorrhagischen Anteilen. Mikroskopisch zeigt der Tumor identische Merkmale zu Manifestationen in anderen Kopf-Hals-Regionen. Dazu gehören irreguläre Nestbildungen und bandartige Anordnungen eosinophiler Zellen, die eine starke Verhornung ausprägen und eine desmoplastische Stromareaktion induzieren. Unterschieden wird zwischen gut, moderat und gering differenzierten verhornenden Plattenepithelkarzinomen.
Therapie der Wahl ist die Tumorresektion mit adjuvanter Radiotherapie. Bei inoperablen Befunden ist eine primäre Radiochemotherapie anzustreben. Die 5-Jahres Gesamt-Überlebensrate für das sinunasale Plattenepithelkarzinom liegt zwischen 50 und 60% und ist stark abhängig vom Tumorstadium bei Diagnosestellung. Karzinome der Nasenhaupthöhle haben eine bessere Prognose als bei Manifestationen innerhalb der Nasennebenhöhlen, da diese erst in größeren Tumorstadien zu Beschwerden führen [217] [217] [219] [224] [225].
2.4.2.1.1.2 Nichtverhornendes Plattenepithelkarzinom
Das nichtverhornende Plattenepithelkarzinom des Sinunasaltrakts wird charakterisiert durch ein ausgeprägtes bandförmiges Wachstumsmuster mit fehlender oder eingeschränkter Reifung.
Synonym werden die Begriffe Schneidersches Karzinom, Zylinderzellkarzinom oder Übergangs-Karzinom verwendet.
Das nichtverhornende Plattenepithelkarzinom repräsentiert ca. 10–27% der sinunasalen Plattenepithelkarzinome. Es betrifft Patienten im 6.-7. Lebensjahrzehnt, darunter deutlich mehr männliche Patienten [226] [226] [227] [229].
Die Risikofaktoren sind ähnlich denen des verhornenden Plattenepithelkarzinoms des Sinunasaltrakts, allerdings können in 30–50% der Fälle transkriptional aktive high-risk HPV Viren nachgewiesen werden. Zwischen 2–10% der sinunasalen Papillome können durch eine maligne Transformation in ein verhornendes und noch seltener in ein nicht verhornendes Plattenepithel übergehen [106] [223].
Makroskopisch zeigt der Tumor ein variables exophytisches und/oder invertiertes Wachstumsmuster mit brüchig wirkender Struktur und Nekrose- sowie hämorrhagischen Arealen. Mikroskopisch wächst der Tumor als sich ausdehnende Nester oder anastomosierende, bandartige Anordnungen von Zellen in der Submukosa mit einer Auflage aus glattem Stroma. Papilläre Eigenschaften können innerhalb oder auf der Oberfläche des Tumors identifiziert werden [106].
Ähnlich dem verhornenden sinunasalen Plattenepithelkarzinom ist die endonasale oder offene Tumorresektion anzustreben, gefolgt von einer adjuvanten Radiotherapie, alternativ zu einer primären Radiochemotherapie für inoperable Befunde. Die 5-Jahres Gesamt-Überlebensrate liegt für nichtverhornende Plattenepithelkarzinome bei ca. 60%. HPV-assoziierte Karzinome haben bessere Überlebenschancen, obwohl die prognostische Signifikanz nicht so stark untermauert ist wie bei Tumoren des Oropharynx [227] [230]. Lymphknotenmetastasen liegen in 3,3 bis 26% der Fälle bei Erstdiagnose vor [231] [232].
2.4.2.1.1.3 Spindelzell-/Sarkomatoides Plattenepithelkarzinom
Das Spindelzell-Plattenepithelkarzinom oder sarkomatoides Karzinom des Sinunasaltrakts ist eine Sonderform des Plattenepithelkarzinoms, die durch das Vorhandensein vorwiegend maligner Spindelzellen und/oder pleomorpher Zellen definiert ist.
Diese Sonderform manifestiert sich vorwiegend bei älteren, männlichen Patienten. Der Tumor tritt äußerst selten im Sinunasaltrakt auf und repräsentiert weniger als 5% aller Plattenepithelkarzinome dieser Region [227] [233] [233] [235].
Die Entstehung des Spindelzell-Plattenepithelkarzinoms ist eng assoziiert mit Tabakkonsum und Strahlenexposition. In den wenigen, bisher bekannten Fällen konnte keine HPV-Infektion nachgewiesen werden [227] [236] [237].
Symptome betroffener Patienten sind zunächst unspezifisch und äußern sich meist in Nasenatmungsbehinderung und Epistaxis. In höheren Tumorstadien können Gesichtsschwellungen und Diplopie sowie Schmerzen der betroffenen Areale hinzukommen [106].
Spindelzell-Plattenepithelkarzinome wachsen als polypöse Raumforderungen mit ulzerierender Oberfläche und ähneln dem makroskopischen Bild der häufiger vorkommenden laryngealen Befunde. Sie stammen vom Plattenepithel ab und zeigen eine abweichende Differenzierung mit epithelial-mesenchymaler Transition auf. Die Tumore können Residuen von dysplastischem Plattenepithel enthalten und zeigen häufig Areale mit Transitionen hin zu malignen Spindel- oder pleomorphen Tumorzellen [106].
Hinsichtlich der Prognose und prädiktiver Faktoren bei sinunasaler Manifestation existieren aufgrund der extrem geringen Fallzahlen keine genaueren Daten.
2.1.2.1.2 Lymphoepitheliales Karzinom
Das lymphoepitheliale Karzinom ist ein gering differenziertes Plattenepithelkarzinom oder ein histologisch undifferenziertes Karzinom. Ein prominentes lymphoplasmatisches Infiltrat, welches dem Nasopharynx-Karzinom ähnelt, ist typisch für das mikroskopische Erscheinungsbild des Tumors.
Die Entität ist sehr selten und tritt - ähnlich dem Nasopharynx-Karzinom – meist in südostasiatischen Ländern auf. Der Altersgipfel liegt zwischen 50 und 70 Jahren, bevorzugt bei Männern (m:w 3:1). Ätiologisch ist bei den lymphoepithelialen Karzinomen eine Assoziation mit dem Epstein-Barr Virus nachweisbar [10] [238].
Manifestationen lassen sich meist in der Nasenhaupthöhle feststellen, die Nasennebenhöhlen sind seltener in den Nasennebenhöhlen verortet. Infiltratives Wachstum in den Gaumen, die Orbita oder die Schädelbasis sind möglich.
Symptome betroffener Patienten sind Druckgefühl, Nasenatmungsbehinderung, Epistaxis und bei Infiltration der Orbita eine Protrusio bulbi. Nervenausfälle können bei intrakranieller Infiltration auftreten [238] [239].
Lymphknoten- und Fernmetastasierung sind möglich. Eine gründliche Endoskopie und Biopsie des Nasopharynx sollten vorgenommen werden, um eine lokoregionäre Ausbreitung eines Nasopharynx-Karzinoms auszuschließen.
Aufgrund der geringen Fallzahl existiert keine Standard-Therapie. Aufgrund der hohen Radiosensitivität wird in den meisten Fällen eine lokoregionäre Radiotherapie durchgeführt, die selbst bei Vorliegen von Halslymphknoten-Mestastasierung ein sehr gutes Ansprechen zeigt. Eine Radiochemotherapie gefolgt von Salvage-Chirurgie ist bei großen Befunden möglich [240]. Die Datenlage ist allerdings aufgrund der sehr geringen Fallzahl schlecht beurteilbar.
2.4.2.1.3 Sinunasales undifferenziertes Karzinom (SNUC)
Das sinunasale undifferenzierte Karzinom wurde erstmals 1986 von Frierson et al. beschrieben [241]. Es handelt sich um ein hochaggressives Karzinom, welches lokal extensives Wachstum zeigt. Pleomorphe Tumorzellen und zahlreiche Tumornekrose-Areale prägen das Erscheinungsbild. Es handelt sich um eine high-grade epitheliale Neoplasie mit unsicherer Histogenese mit oder ohne neuroendokriner Differenzierung. Eine genaue Abgrenzung hinsichtlich des lymphoepithelialen Karzinoms und des Olfaktoriusneuroblastoms ist entscheidend [10] [241].
Der Altersbereich liegt zwischen 30 und 90 Jahren mit einer höheren Inzidenz bei Männern (M:W 2–3:1) [238] [242].
Es existiert keine Assoziation mit dem Epstein-Barr Virus. Einige Fälle traten nach vorangegangener Radiotherapie im Rahmen eines Nasopharynx-Karzinoms auf [238].
Das SNUC manifestiert sich meist in der Nasenhaupthöhle, dem Sinus maxillaris und ethmoidalis mit häufiger Infiltration benachbarter Strukturen. So zeigen bis zu 50% der Patienten zum Zeitpunkt der Diagnosesicherung bereits Infiltrationen der Dura und 30% Infiltrationen der Orbita [243] [244]. In einer Studie, welche die prozentuale Häufigkeit des T-Stadiums in Relation zu den untersuchten Karzinomhistologien analysierte, zeigten sich für das SNUC 69% der Patienten im Stadium T4 [245].
Ähnlich dem lymphoepithelialen Karzinom kommt es bei betroffenen Patienten initial zu Nasenatmungsbehinderung, Epistaxis, später zu Protrusio bulbi, periorbitalen Schwellungen sowie Gesichtsschmerzen und bei intrakranieller Infiltration zu Ausfällen von Hirnnerven.
Das SNUC zeigt eine hohe Rate von Lokalrezidiven und Fernmetastasierung mit häufiger Gefäß- und Nerveninfiltration [246] [247]. Die 5-Jahres Überlebensrate liegt laut einer aktuellen Studie, die insgesamt 318 Patienten mit SNUC in den USA analysiert hat, bei 34,9% und die 10-Jahres Überlebensrate bei 31,3% [248]. Die schlechte Prognose ist in den meisten Fällen durch das bereits hohe Tumorstadium zum Zeitpunkt der Diagnosestellung und die hierdurch bedingte Inoperabilität verursacht. Ein weiterer Grund ist die Tatsache, dass die Mehrzahl der publizierten Fallserien weniger als 20 Patienten enthalten, weshalb kein standardisierter Therapieansatz oder eine Leitlinie zur Therapie des SNUC existiert [243].
In Anbetracht der aktuellen Literatur besteht ein Konsens, dass eine multimodale, aggressive Therapie aus einer Kombination von Chirurgie, Radio- und Chemotherapie erfolgen sollte. Dies wurde durch 2 große SEER (Surveillance, Epidemiology and End Results Program) Datenbank Studien untermauert [248] [249]. Die Resektion des Tumors gefolgt von einer adjuvanten Radiochemotherapie oder die primäre Radiochemotherapie sind mögliche Therapieansätze in Abhängigkeit der Resektabilität des Tumors. Ansätze einer Targeted Therapy mit Lapatinib, welches den Human epidermal growth factor receptor 2 (HER 2) Singalweg sowohl in vitro als auch in vivo unterdrückte, zeigten vielversprechende erste Ergebnisse, die auf effektivere Therapien hoffen lassen [250].
2.4.2.1.4 Adenokarzinom
2.4.2.1.4.1 Adenokarzinom vom intestinalen Typ
Sinunasale Adenokarzinome vom intestinalen Typ ähneln dem morphologischen Aufbau des Adenokarzinoms des Darms.
Das Auftreten ist sehr selten und wird auf eine Inzidenzrate von weniger als 1:1 000 000 Personenjahren geschätzt. Allerdings variiert die Inzidenz deutlich und steigt hinsichtlich der Prävalenz um den Faktor 500 bei Mitarbeitern von Holz- und Leder-verarbeitenden Betrieben. Das Karzinom ist hierbei eine anerkannte Berufskrankheit (BK4203). Männer sind 3–4 mal häufiger als Frauen betroffen, was vermutlich auf die Beschäftigungsverhältnisse in den entsprechenden Berufen zurückzuführen ist. Das Alter betroffener Patienten liegt meist zwischen dem 6. und 7. Lebensjahrzehnt [106].
Manifestationen sind häufig nahe der lateralen Nasenwand und der mittleren Nasenmuschel zu finden. Schätzungen zufolge entwickeln sich 40% der Tumore im Sinus ethmoidalis, 28% in der Nasenhaupthöhle und 23% im Sinus maxillaris [251] [252].
Symptome sind unilaterale Nasenatmungsbehinderung, Epistaxis und Rhinorrhoe. Seltener treten Schmerzen, Gesichtsschwellungen und Diplopie oder eine Protrusio bulbi auf. Eine Destruktion des umgebenden Knochens und das Einwachsen in benachbarte Regionen ist häufig [106] [252].
Makroskopisch zeigt sich eine polypoide, teilweise papillär und nodulär anmutende Raumforderung mit brüchiger Konsistenz und ulzerierenden, hämorrhagischen sowie seltener gelatineartigen oder mukösen Anteilen [106] [251] [252]. Mikroskopisch lässt sich exophytisches, papilläres und tubuläres oder muzinöses oder vorwiegend aus Siegelringzellen bestehendes Wachstum feststellen. Der Grad der Differenzierung ist stark variabel. In 6–10% kann eine KRAS Mutation festgestellt werden. Eine BRAF-Mutation ist mit<10% deutlich seltener [253] [253] [254] [256].
Die radikale Tumorresektion mit freien Resektionsrändern ist aufgrund der eher schlechten Strahlensensibilität der Tumore Therapie der Wahl. Durch die Weiterentwicklung der endoskopischen Technik ist die transnasale Resektion in einigen Fällen auch bei orbitaler und intrakranieller Infiltration möglich [257], mitunter unter Berücksichtigung komplexer rekonstruktiver Verfahren [258]. Low-grade Adenokarzinome vom intestinalen Typ mit niedrigem Tumorstadium können durch eine radikale Tumorresektion alleine therapiert werden. Bei allen anderen Stadien und Entitäten sollten eine adjuvante Radiotherapie erfolgen [259]. Die intensitätsmodulierte Photonenbestrahlung kann als Teil des Therapieregimes diskutiert werden [260]. Bislang existieren keine Phase III Studien hinsichtlich einer Chemotherapie, weshalb die aktuellen Ergebnisberichte hauptsächlich auf Fallberichten und kleinen retrospektiven Fallserien basieren. Im Rahmen einer prospektiven Phase II Studie konnte bei neoadjuvanter Chemotherapie mit PFL (Cisplatin, 5-Fluoruracil und Leucovorin) bei 16% der behandelten Patienten eine primäre Komplettremission gezeigt werden [261]. In diesen Fällen lag das 3-Jahres Gesamtüberleben bei 100% gegenüber 43% bei den restlichen Patienten.
Low-grade papilläre Adenokarzinome haben die besten 3-Jahres Überlebensraten mit<80 und<60% nach 5 Jahren. Tumore der Grade 2 und 3 weisen 3-Jahres Überlebensraten von 54 bzw. 35% auf. Muzinöse Tumore mit alveolärem Wachstum haben ähnliche Überlebensraten wie papilläre Tumore des Grades 2. Das aggressivste Wachstum weisen Siegelring-Adenokarzinome auf. Bei Infiltration der Orbita, der Haut, des Sinus ethmoidalis oder sphenoidalis sowie der Schädelbasis ist die Prognose deutlich schlechter. Lymphknotenmetastasen können bei Diagnosestellung in 8% und Fernmetastasen in 13% der Fälle diagnostiziert werden [106] [252].
2.4.2.1.4.2 Adenokarzinom vom nicht intestinalen Typ
Das sinunasale Adenokarzinom vom nicht intestinalen Typ zeigt keine Eigenschaften von Speicheldrüsenkarzinomen und besitzt keinen intestinalen Phänotyp. Die genannte Tumorkategorie ist heterogen. Trotzdem weist sie spezifische Entitäten auf, die einzigartig sind (z. B. das Nierenzell-ähnliche Karzinom) [106].
Sinunasale low-grade nicht intestinale Adenokarzinome kommen sehr selten vor, weisen keine Prädilektion eines spezifischen Geschlechts auf und können innerhalb einer Altersspanne von 9–89 Jahren diagnostiziert werden. Das mittlere Alter bei Diagnosestellung liegt innerhalb des 6. Lebensjahrzehnts. High-grade nicht intestinale Adenokarzinome sind selten, betreffen häufiger männliche Patienten, weisen ebenfalls eine breite Altersspanne mit einem Altersgipfel innerhalb des 6. Lebensjahrzehnts auf. Weder für low-grade noch für high-grade nicht intestinale Adenokarzinome des Sinunasaltrakts ist die Ätiologie bekannt.
Am häufigsten manifestieren sich die Tumore in der Nasenhaupthöhle (64%), gefolgt vom Sinus ethmoidalis (20%). Ca. die Hälfte aller high-grade nicht intestinale Adenokarzinome weisen bei Diagnosestellung bereits ein fortgeschrittenes Tumorstadium mit Infiltration der Nasenhaupthöhle und Nasennebenhöhlen auf [262] [262] [263] [265].
Symptome der low-grade Variante sind häufig unspezifisch und beinhalten Nasenatmungsbehinderung, Epistaxis und Schmerzempfindungen. High-grade Tumore verursachen häufiger Deformitäten der Gesichtshaut und Protrusio bulbi bei orbitaler Infiltration. Bildmorphologisch präsentieren sich low-grade nicht intestinale Adenokarzinome als solide, die Nasenhaupthöhle oder Nebenhöhlen ausfüllende Masse, wohingegen high-grade nicht intestinale Adenokarzinome deutlich destruktiveres Wachstum mit ossärer Infiltration und Invasion der umgebenden anatomischen Areale zeigen [106].
Makroskopisch imponieren die low-grade Tumore als rote, polypoide oder himbeerartig konfigurierte, feste Struktur. Histologisch zeigen sich papilläre und/oder tubuläre Eigenschaften mit komplexem Wachstumsmuster. Eine einzelne Schicht uniformer muzinöser, kuboider bis säulenförmiger Epithel-Zellen umgibt die Tumore. High-grade Tumore zeigen deutlich mehr histologische Diversität. Solides Wachstum mit gelegentlich eingestreuten glandulären Strukturen sowie vereinzelten Mukozyten sind typisch. Einige Varianten zeigen nestartige Verteilungsmuster mit infiltrativem Verhalten. Zahlreiche Mitoseformen und Nekroseareale sind vorhanden [106].
Therapie der Wahl ist die radikale Tumorresektion. Trotz der spärlichen Datenlage aufgrund der wenigen beschriebenen Fälle erscheint die alleinige chirurgische Therapie – tumorfreie Resektionsränder vorausgesetzt – prognosebestimmend zu sein [266]. Eine kürzlich durchgeführte National Cancer Database Analyse zeigte keinen Einfluss einer Radiotherapie auf das Überleben von Patienten mit low-grade Adenokarzinomen [267]. Patienten mit high-grade Adenokarzinomen zeigen eine deutlich bessere Überlebensrate bei multimodaler Therapie inklusive radikaler Tumorresektion und adjuvanter Radiotherapie. Die Rolle einer systemischen Therapie konnte aufgrund der geringen Fallzahlen bislang nicht abschließend geklärt werden [257].
Rezidive sind bei 25% der low-grade Variante zu finden und nur etwa 6% der betroffenen Patienten sterben an der Erkrankung. Die Prognose für Patienten mit high-grade nicht intestinalem Adenokarzinom ist deutlich schlechter. Die meisten Patienten versterben innerhalb von 5 Jahren nach Diagnosestellung an der Erkrankung. Lokale und Fernmetastasierung tritt gelegentlich auf [262] [262] [264].
Bei den wenigen berichteten Fällen eines Nierenzell-ähnlichen Karzinoms gab es weder Rezidive noch Metastasierungen [268].
2.4.2.1.5 Speicheldrüsenartige Karzinome
Speicheldrüsen-Neoplasien in der Nase oder den Nasennebenhöhlen sind extrem selten. Die überwiegende Mehrzahl der Fälle besteht aus malignen Entitäten. In einer Analyse von Daniel K. Heffner zeigte sich bei insgesamt 311 Patienten mit sinunasalen speicheldrüsenartigen Tumoren folgende Verteilung der Entitäten [269]:
In der folgenden Darstellung werden die malignen sinunasalen Entitäten dargestellt:
-
Adenoidzystisches Karzinom
-
Azinuszellkarzinom
-
Mukoepidermoidkarzinom
-
Epithelial-myoepitheliales Karzinom
2.4.2.1.5.1 Adenoidzystisches Karzinom (ACC)
Das adenoidzystische Karzinom tritt v. a. in den großen und kleinen Speicheldrüsen auf. Eine Manifestation ist aber in allen Bereichen auftreten, in denen sekretorische Drüsen lokalisiert sind (Mamma, Cervix, Colon, Prostata, Nasenhaupthöhle und Nasennebenhöhlen). Es ist das häufigste speicheldrüsenartige Malignom des Sinunasal-Trakts mit einer Altersspanne von 11–92 Jahren. Sinunasale Manifestationen finden sich im Sinus maxillaris (60%) und in der Nasenhaupthöhle (25%). Adenoidzystische Karzinome repräsentieren 5% aller sinunasalen malignen Neoplasien mit einer Inzidenz von 25% bezogen auf alle Speicheldrüsenkarzinome [270] [270] [271] [272] [274]. Es wächst langsam, aber infiltrativ entlang beteiligter Hirnnerven in umgebende Strukturen ein. Hämatogene Metastasierung ist häufig und kann selbst Jahre nach der Erstdiagnose auftreten. Aufgrund des späten Auftretens von Symptomen weisen die betroffenen Patienten häufig hohe Tumorstadien auf, was zu entsprechender Morbidität bei Tumorresektionen oder primärer Radiotherapie führt. Aufgrund der geringen Strahlensensitivität sind Dosen>80 Gy erforderlich, was zu Kollateralschäden benachbarter Strukturen (Orbita, Nervus opticus, Cerebrum) führt [275] [275] [277].
Therapie der Wahl ist die radikale Tumorresektion und postoperative Radiotherapie für R1/R2 Resektionen, bei perinodaler Infiltration und in fortgeschrittenen Tumorstadien (T3/T4), obwohl einige Studien für alle Tumorstadien (T1-4) ein besseres Outcome nachweisen konnten [275] [278] [278] [279] [281].
Aufgrund der hohen Tumorstadien bei Erstdiagnose und der häufigen Spätrezidive ist die Prognose des adenoidzystischen Karzinoms schlecht. Die 5-Jahres Überlebensrate wird mit 38–64% bei konventioneller Bestrahlung angegeben. Die lokale Tumorkontrolle erreichte bei Anwendung von Protonenbestrahlung 50–70% und sogar bis zu 93% bei Neutronenbestrahlung [272] [275] [276] [282] [282] [283] [284] [286].
2.4.2.1.5.2 Azinuszellkarzinom
Das Azinuszellkarzinom ist ein seltenes Malignom der Speicheldrüsen, welches vorwiegend in der Glandula parotis anzutreffen ist. Nach Tumorresektion mit oder ohne postoperativer Radiotherapie werden 20-Jahres Überlebensraten nahe 90% erreicht [287]. Eine Manifestation im Sinus maxillaris oder ethmoidalis, an den Nasenmuscheln oder dem Nasenseptum ist außerordentlich seltenen. Die derzeitige englischsprachige Literatur beschreibt 19 Fälle, deren Follow Up von 1 bis 22 Jahren reicht. 5-Jahres und 10-Jahres Überlebensraten lieben über 90% [288] [288] [289] [291].
2.4.2.1.5.3 Mucoepidermoidkarzinom
Sinunasale Mucoepidermoidkarzinome sind Malignome der kleinen Speicheldrüsen, die sich in der Mundhöhle und dem Oropharynx manifestieren und an diesen Manifestationsorten eine 5-Jahres Überlebensrage von 87% aufweisen [292]. Lymphknotenmetastasten treten in bis zu 15% der Fälle auf.
Sinunasale Mucoepidermoidkarzinome repräsentieren ca. 1,5% aller sinunasalen Malignome und sind damit sehr selten. Häufigste Manifestationsorte sind Sinus maxillaris und die Nasenhaupthöhle [293], in denen sich in 46% high grade Mucoepidermoidkarzinome diagnostizieren lassen. Symptome befallener Patienten sind Nasenatmungsbehinderung, chronisch sinunasale Beschwerden, Gesichtsschmerzen, Epistaxis und Cephalgien. Kleine monozentrische Studien zeigten 5-Jahres Überlebensraten von 35,9% bis 44,1% bei sinunasaler Manifestation [294] [295]. Die Tumorresektion mit adjuvanter Radiatio ist Therapie der Wahl, allerdings fehlen aufgrund der geringen Fallzahlen Daten hinsichtlich der Effektivität [293].
2.4.2.1.5.4 Epithelial-myoepitheliales Karzinom
Das epithelial-myoepitheliale Karzinom repräsentiert eine außerordentlich seltene maligne Entität, welche sich vorwiegend in den großen Speicheldrüsen manifestiert und ca. 1% aller Speicheldrüsenmalignome ausmacht. Noch seltener ist die Manifestation in der Nasenhaupthöhle oder den Nasennebenhöhlen, von der nur wenige Fallberichte existieren [296]. Die größte Kohortenstudie umfasst 468 Patienten mit epithelial-myoepithelialem Karzinom. 18 dieser Patienten zeigten eine Manifestation im sinunasalen Bereich [297]. 80% der Patienten waren älter als 50 Jahre alt. Weibliche Patienten waren häufiger betroffen (W:M 1,5–6:1) [297]. Aufgrund der niedrigen Fallzahlen ist die Beschreibung der Pathophysiologie und der Therapiestrategien nicht systematisiert, da ein großer Teil der relevanten Literatur aus Fallberichten besteht.
Der Begriff epithelial-myoepitheliales Karzinom ist ein histopathologischer Terminus, der die Proliferation tubulärer Strukturen mit einer zweischichtigen Zellauskleidung beschreibt. Die innere Schicht besteht aus kubischen oder niedrig zylindrischen Duktalzellen, die äußere Schicht aus hellen epitheloiden Zellen. Das Tumorstruma kann hyalinisiert sein [298].
Je nach Lokalisation präsentieren die Patienten Symptome wie Epistaxis, Druckgefühl und Schmerzen, Schwellungen und Rhinorrhoe. Häufig zeigt sich bei der Endoskopie eine hämorrhagisch anmutende Raumforderung. Computertomografisch ist eine heterogen Kontrastmittel-aufnehmende Weichteil-Struktur in den beteiligten Nasennebenhöhlen darstellbar. Ossäre Destruktionen benachbarter Strukturen sind nicht typisch, können aber auftreten [296].
Aktuelle Berichte zeigten einen nur geringen Glukose-Uptake in der PET-CT, was dem low-grade Potenzial der Malignität zuzuordnen ist und eine präoperative, bildgebende Hilfestellung hinsichtlich der Diagnose erschwert [299] [300].
Therapie der Wahl ist die großzügige chirurgische Exzision. Die Effektivität einer adjuvanten Radiotherapie ist aufgrund des inhomogenen biologischen Verhaltens und Ansprechens unklar [301].
Lymphknotenmetastasen sind selten und treten in weniger als 5% der Fälle auf. Die 5- und 10-Jahres Überlebensrate liegt bei 72,7 und 59,5% [297].
2.4.2.2 Neuroendokrine Neoplasien
Der Begriff Neuroendokrine Neoplasien umfasst mehrere Entitäten. Neben dem neuroendokrinen Karzinoid und dem klassischen neuroendokrinen Karzinom zählen auch Aesthesioneuroblastome (siehe 2.4.2.6.2) sowie sinunasale undifferenzierte Karzinome (SNUC, siehe 2.4.2.1.3) zu den Neoplasien mit neuroendokriner Differenzierung [302].
Neuroendokrine Neoplasien exprimieren neuroendokrine Marker wie Synaptophysin und Chromogranin A. Große membrangebundene (hormonenthaltende) Vesikel sind charakteristische Merkmale. Epitheliale neuroendokrine Karzinome (gut, mittelgradig oder schlecht differenziertes Karzinoid) bilden sich aus Zellen des diffusen neuroendokrinen Zellsystems, die Zytokeratine exprimieren und in der Schleimhaut lokalisiert sind. Olfaktoriusneuroblastome und Paragangliome hingegen sind neuroektodermaler Herkunft und bilden sich aus der Olfaktoriusmembran bzw. den Kopf- Hals-Paraganglien [303]. Entsprechend der WHO Klassifikation werden neuroendokrine Tumore als Neoplasie Grad I (früher low-grade neuroendokriner Tumor oder typisches Karzinoid), Neoplasie Grad II (früher intermediärer neuroendokriner Tumor oder atypisches Karzinoid) und Neoplasie Grad III, kleinzelliges Karzinom (früher high-grade Neuroendokriner Tumor oder kleinzelliges Karzinom) sowie Neoplasie Grad III, großzelliges Karzinom (früher high-grade neuroendokriner Tumor oder großzelliges neuroendokrines Karzinom) eingeteilt [304].
Neuroendokrine Karzinoide (typisches Karzinoid, Neoplasie Grad I)
Über 90% der neuroendokrinen Karzinoide manifestieren sich am Larynx (supraglottisch). Sinunasale Manifestationen sind wesentlich seltener und führen zu gut abgrenzbaren, submukösen und häufig polypoiden Tumoren [305]. Vor allem schlecht differenzierte neuroendokrine Karzinome finden sich in der Nasenhaupthöhle, den Nasennebenhöhlen oder an der Schädelbasis. Die 5-Jahres Überlebensrate liegt bei 5–20% [106].
Die Symptome hängen vom Manifestationsort ab. Ein Karzinoid-Syndrom ist bei übermäßiger Hormonproduktion möglich, vor allem bei hepatischer Metastasierung und tritt in etwa 10% aller Patienten mit Karzinoiden auf. Die typische Trias beinhaltet anfallsartige Rötung von Gesicht und Oberkörper, Diarrhoe und kardiale Beteiligung [306].
Neuroendokrine Karzinome
Das sinunasale neuroendokrine Karzinom ist ein high-grade Karzinom mit morphologischen und immunhistochemischen Merkmalen einer neuroendokrinen Differenzierung [106]. Es wird unterteilt in kleinzellige neuroendokrine Karzinome und großzellige neuroendokrine Karzinome und stellt ca. 3% aller sinunasalen Tumore und tritt häufiger bei Männern mittleren bis höheren Alters (mittleres Alter 49–65 Jahre) auf. In seltenen Fällen wird über eine Assoziation mit High-Risk humanen Papilloma-Viren berichtet.
Primärer sinunasaler Manifestationsort ist der Sinus ethmoidalis, die Nasenhaupthöhle, gefolgt von den Sinus maxillaris und sphenoidalis.
Symptome einer Manifestation sind weitestgehend unspezifisch (Nasenatmungsbehinderung, Rhinorrhoe und chronische sinunasale Beschwerden). Viele Patienten werden erst in höheren Tumorstadien bei ihrem behandelnden Arzt vorstellig.
Endoskopisch zeigt sich eine meist große, hämorrhagische Tumormasse mit nekrotischen Anteilen. Bildmorphologisch zeigen sich ossäre Destruktionen und Infiltrationen benachbarter anatomischer Regionen.
Histopathologisch ist das sinunasale neuroendokrine Karzinom identisch zu den in Lunge und anderen Kopf-Hals-Lokalisationen vorkommenden neuroendokrinen Karzinomen. Es zeigt sich ein hochgradig infiltratives Wachstum mit häufiger perineuraler und lymphovaskulärer Infiltration. Sinunasale neurendokrine kleinzellige und großzellige Karzinome weisen eine Zytokeratin-Expression auf, was eine Differenzierung z. B. gegenüber dem Olfaktoriusneuroblastom ermöglicht [304].
Therapeutisch wird – sofern möglich – eine Tumorresektion gefolgt von adjuvanter Radiochemotherapie angestrebt. Vor allem für das großzellige neuroendokrine Karzinom scheint eine neoadjuvante Radiochemotherapie gefolgt von einer Tumorresektion bessere Ergebnisse zu erzielen [302].
Die 5-Jahres Überlebensrate beträgt 50–65%. Eine Lokalisation im Sinus sphenoidalis ist prognostisch günstiger (~80%) als eine maxilläre oder ethmoidale Manifestation (~33%). Trotz eingeschränkter Datenlage aufgrund der geringen Fallzahlen scheint die Prognose für großzellige neuroendokrine Karzinome günstiger zu sein als für die kleinzellige Variante [106].
2.4.2.3 Maligne Weichteiltumore (Sarkome)
2.4.2.3.1 Fibrosarkom
Das Firbosarkom ist ein maligner spindelzelliger Tumor mit faszikulärer Architektur und variabel ausgeprägter Collagenmatrix-Produktion, welche fibroblastäre/myofibroblastäre Differenzierung aufweist. Weiter zeigt das Fibrosarkom eine niedrige Mitoseraten und seltene nukleäre Pleomorphismen oder anaplastische Eigenschaften.
Manifestationsorte sind meist in den Extremitäten lokalisiert, lediglich 1% der Fibrosarkome ist im Kopf-Hals-Bereich zu finden. Es ist für weniger als 3% aller nichtepithelialen Malignome des Kopf-Hals-Bereichs verantwortlich, stellt allerdings die zweithäufigste Entität von Kopf- Hals-Sarkomen dar [106] [307].
Symptome ähneln denen anderer Neoplasien der Nasenhaupthöhle und Nasennebenhöhlen. Unspezifische Beschwerden wie Nasenatmungsbehinderung und Epistaxis, Druckgefühl, Schmerzen und Schwellungen können auftreten.
Das Fibrosarkom hat ein hohes Lokalrezidiv-Risiko, allerdings ein geringes Risiko für Fernmetastasierung [140] [308]. Trotz der sehr geringen Fallzahlen wird eine großzügige Tumorresektion empfohlen, da ein geringer Sicherheitsabstand das Risiko des Lokalrezidivs unverhältnismäßig erhöht [140]. Eine U.S. National Cancer Instituteʼs Surveillance, Epidemiology, and End Results (SEER) Datenbankanalyse untersuchte den Verlauf von 51 Patienten mit sinunasalen Fibrosarkomen über einen Zeitraum von 1973 bis 2012 [307]. Das mittlere Alter lag bei 54.5 Jahren ohne Überwiegen eines Geschlechts. Interessanterweise waren 83,7% der Patienten hellhäutig und nur 8,2% dunkelhäutig. Die häufigste Lokalisation war mit 54,9% der Sinus maxillaris gefolgt von der Nasenhaupthöhle (23,5%). Der häufigste histologische Typ war das mittelgradig differenzierte (59,5%), gefolgt vom gut differenzierten Fibrosarkom (16,2%). Im Follow up zeigte sich bei 28,2% der Patienten ein Lokalrezidiv, in 64,1% kam es zu einer regionalen Metastasierung und 7,7% wiesen Fernmetastasen auf.
Häufigste Therapiemodalität war die alleinige Tumorresektion (61,2%), gefolgt von Tumorresektion mit adjuvanter Radiotherapie (32,7%). Auch Lokalrezidive wurden primär durch erneute Tumorresektion therapiert (71,4%) gefolgt von Tumorresektion mit adjuvanter Radiotherapie (7,1%). Die 5-Jahres Überlebensrate lag für alle nachverfolgbaren Fälle bei 71,7%. Aktuelle Studien zeigen deutliche Vorteile bei Patienten, die eine adjuvante Radiotherapie erhalten [308] [309].
2.4.2.3.2 Undifferenziertes pleomorphes Sarkom/Malignes fibröses Histiozytom
Das undifferenzierte pleomorphe Sarkom (Synonym: malignes fibröses Histiozytom) ist ein high-Grade Weichteilsarkom ohne Differenzierungslinie. Es tritt bei Erwachsenen auf, eine sinunasale oder Schädelbasis-Manifestation ist sehr selten, obwohl es nach dem Rhabdomyosarkom und dem Fibromyosarkom das dritthäufigste Sarkom in diese Lokalisationen darstellt [106].
Vorangegangene Radiotherapien werden für das Entstehen des undifferenzierten pleomorphen Sarkoms verantwortlich gemacht [310] [311].
Vor allem unspezifische Symptome wie schmerzlose Schwellungen, Nasenatmungsbehinderung, Protrusio bulbi und Diplopie sowie Epistaxis können auftreten. Eher selten kommt es zu regionaler oder Fernmetastasierung [106].
Endoskopische zeigt sich eine lobulierte, gräulich-weißliche, teils fleischige Masse mit hämorrhagischen Anteilen. Die meisten Befunde erscheinen umschrieben. Mikroskopisch präsentiert das Malignom Spindel- und pleomorphe Zellen in einer variabel kollagenisierten, extrazellulären Matrix. Pleomorphismen, atypische Mitosen, Tumornekroseareale, Histozytenartige und Schaumzellen zeigen sich häufig. Viele Tumorzellen weisen Eigenschaften von Fibroblasten, Myofibroblasten oder Histiozyten auf.
Das undifferenzierte pleomorphe Sarkom ist eine Ausschlussdiagnose, welche nach Ausschluss von Schleimhautmelanomen, Karzinomen, Lymphomen und anderen Sarkomen gestellt werden kann.
Die Tumorresektion scheint trotz der geringen Datenlage und unabhängig von der Randsituation essentiell zu sein. Eine Radiotherapie erhöht die Chance auf lokale Tumorkontrolle [106] [311]. Die 5-Jahres Überlebensrate liegt zwischen 60 und 70%.
Gerrand et al. zeigten für Sarkome der Extremitäten in einer Studie ein unterschiedliches Outcome für Patienten abhängig von histologischem Subtyp, der Anwendung einer Radiotherapie, der lokalen Anatomie und ungeplanter Exzision vor Überweisung in ein Zentrum [312] und postulierten, Patienten bei dem Verdacht auf ein Weichteilsarkom in ein spezialisiertes Diagnostik- und Therapie-Zentrum mit multidisziplinären Sarkom-Spezialisten zu überweisen [311].
2.4.2.3.3 Leiomyosarkom
Das Leiomyosarkom ist ein maligner Tumor, der von der glatten Muskulatur ausgeht. Typische Manifestationsorte sind Uterus oder Intestinaltrakt. Die sinunasale Manifestation oder Lokalisation an der Schädelbasis sind extrem selten. Betroffen sind vorwiegend Erwachsene, nur in Ausnahmefällen Kinder. Auch für das Leiomyosarkom spielt eine vorangegangene Radiotherapie eine große Rolle für die Genese [106].
Aufgrund nur unspezifischer Symptome werden die Patienten meist erst in späten Tumorstadien bei ihrem behandelnden Arzt vorstellig, was zu schlechteren Prognosen bei sinunasalen Manifestation im Vergleich zu anderen Lokalisationen führt [313]. Klinisch zeigt sich eine weiche, vorwiegend polypöse Tumormasse, die Schmerzen, Nasenatmungsbehinderung und Epistaxis verursachen kann. Die Läsionen können auch den kraniofazialen Knochen betreffen und je nach Infiltration weiterführende Beschwerden wie Diplopie, Protrusio bulbi, usw. verursachen.
Eine hämatogene Metastasierung in Lunge, Leber, andere Weichteilareale, Knochen oder zerebrale Strukturen sind möglich. Metastasen anderer Regionen sollten vor der definitiven Tumortherapie ausgeschlossen werden.
Makroskopisch ist die Tumormasse polypoid. Sowohl klare Abgrenzungen zum umgebenden Gewebe als auch schwer abgrenzbare Befunde sind möglich. Mikroskopisch zeigt sich infiltratives Wachstum oder scharf demarkierte Grenzen. Spindelzellen, die in vernetzten Faszikeln angeordnet sind, prägen das Erscheinungsbild. Das eosinophile Zytoplasma zeigt oft kleine perinukleäre Vakuolen [106].
Computertomografische Darstellungen sind häufig unspezifisch und zeigen expansive zystische oder nekrotische, heterogene Läsionen innerhalb des Weichgewebes. Im MRT zeigt sich in der T1- und T2-Wichtung nur moderate Hyperintensität, was die Diagnosefindung erschwert [314].
Die Tumorresektion ist Methode der Wahl, allerdings ist aufgrund der Lokalisation und vitaler benachbarter Strukturen eine Resektion mit großem Sicherheitsabstand nur eingeschränkt möglich. Adjuvante Chemotherapie und/oder Radiotherapie kommen bei Patienten mit lokal fortgeschrittenem Wachstum, Rezidiven oder Metastasierungen zum Einsatz [313] [315]. Circa ein Drittel der Patienten mit sinunasaler Manifestation stirbt entweder an Fernmetastasierung oder Lokalrezidiven, die in vitale benachbarte Strukturen einwachsen [106].
2.4.2.3.4 Rhabdomyosarkom
Rhabdomyosarkome sind maligne mesenchymale Tumore mit Skelettmuskeldifferenzierung.
Unterschieden werden
-
embryonale Rhabdomyosarkome
-
alveoläre Rhabdomyosarkome
-
pleomorphe Rhabdomyosarkome
-
spindelzellige Rhabdomyosarkome
Die Begriffe Myosarkom und malignes Rhabdomyom werden synonym mit Rhabdomyosarkom verwendet.
Die Inzidenz von sinunasalen Rhabdomyosarkomen beträgt 0,034:100 000 mit primärer Manifestation in den Nasennebenhöhlen gefolgt von der Nasenhaupthöhle. Es ist das häufigste sinunasale Sarkom sowohl bei Kindern als auch bei Erwachsenen. Der Altersgipfel liegt innerhalb des ersten Lebensjahrzehnts [106] [311] [316]. Eine strahleninduzierte Genese wird diskutiert.
Makroskopisch zeigt sich eine polypoide, schlecht abgrenzbare Tumorformation mit Ausdehnung in benachbarte Strukturen und fleischiger, gelatineartiger, bräunlich bis grauer Oberfläche.
Das embryonale Rhabdomyosarkom ist die am häufigsten im Sinunasaltrakt vorkommende Entität. Sie weist primitive bis spindelförmige Zellen mit spärlichem Zytoplasma und hyperchomatischen Nuklei sowie verstreute Rhabdomyoblasten mit deutlich eosinophilem Zytoplasma auf. Die Anzahl der Rhabdomyoblasten steigt typischerweise nach Radiotherapie deutlich an.
Bei Erwachsenen findet sich häufiger das sinunasale alveoläre Rhabdomyosarkom [311], welches fibrovaskuläre Septen aufweist, die Herde von runden, neoplastischen Zellansammlungen voneinander trennen. Riesenzellen mit mehreren, peripher lokalisierten Zellkernen können präsent sein [106].
Das Outcome der Patienten scheint für Patienten, bei denen eine Vorabexzision oder Probeexzision zur Histologie-Sicherung durchgeführt wurde, schlechter zu sein als für Patienten, bei denen unmittelbar eine vollständige Tumorresektion durchgeführt wurde [311] [312]. Die Überweisung bei Verdacht auf ein Sarkom in ein auf Sarkome spezialisiertes Zentrum mit interdisziplinärer Zusammenarbeit erscheint deshalb besonders wichtig.
Therapeutisch ist die Tumorresektion mit weitem Sicherheitsabstand anzustreben. Auch für die unvollständige Tumorresektion scheint die chirurgische Resektion ein prädiktiver Faktor zu sein [311]. Die Kombination von Operation, Chemotherapie und Bestrahlung werden 5-Jahres Überlebensraten von 40–45% berichtet, die für Patienten unter 18 Jahren und bei weiblichem Geschlecht etwas höher liegen. Bei Infiltration der Schädelbasis ist die Prognose deutlich schlechter [106] [311] [317].
2.4.2.3.5 Angiosarkom
Das Angiosarkom ist eine vaskuläre, maligne Neoplasie. Synonyme Begriffe sind epitheloides Hämangioendoetheliom, malignes Hämangioendotheliom, malignes Angioendotheliom oder Hämangiosarkom.
Der Tumor entwickelt sich in über der Hälfte der Fälle innerhalb der Haut und oberflächlichen Gewebsschichten des Kopf-Hals-Bereichs. Epidemiologisch ist das sinunasale Angiosarkom für weniger als 0,1% aller Kopf-Hals-Malignome und weniger als 1% aller sinunasalen Malignome verantwortlich [106].
Als mögliche, allerdings selten berichtete Risikofaktoren für die Entstehung werden Strahlenexposition, Vinylchlorid- und Kohlestaubexposition diskutiert [318] [318] [320].
Das Angiosarkom ist eher in der Nasenhaupthöhle und dem Sinus maxillaris lokalisiert. Dort führt es zu initial unspezifischen Beschwerden, allen voran Epistaxis und Nasenatmungsbehinderung. Einige Patienten berichten zudem über sinunasale Beschwerden, Epiphora, Schmerzen und Druckgefühl.
Häufig ist bei Angiosarkomen eine Knocheninfiltration zu beobachten, die sich in der Computertomografie darstellen lässt. In der MRT-Darstellung ist in der T2-Wichtung eine kräftige Signalintensität darstellbar. Die präoperative Angiografie kann hilfreich sein, um zuführende Gefäße darzustellen und eine präoperativ Embolisation zu ermöglichen [106] [321]. Lymphknoten- und Fernmetastasierung sind bei Erstmanifestation ungewöhnlich.
Makroskopisch präsentiert sich das Angiosarkom mit einer nodulär bis polypoiden, weichen und lividen bis rötlichen Oberfläche mit Ulzerationen, die zu Blutungen neigen. Mikroskopisch entwickelt sich das Malignom unter einer intakten Epithelschicht mit neoplastischen Gefäßbildungen, die sich in das Weichgewebe und den angrenzenden Knochen erstrecken. Hämorrhagische und nekrotische Areale begleiten das Erscheinungsbild. Innerhalb der Neubildungen zeigen sich kapilläre, kavernöse und rudimentär angelegte Gefäße, die mit Erythrozyten angefüllt und mit vergrößerten, atypischen spindelförmigen oder epitheloiden Endothelzellen ausgekleidet sind. Ein Grading wird für das Angiosarkom nicht vorgenommen [106] [321].
Aufgrund der sehr niedrigen Inzidenz existiert keine Standardtherapie für das sinunasale Angiosarkom. In den meisten Fällen ist die Tumorresektion gefolgt von einer Radiotherapie beschrieben. Weitere Therapieansätze, die in einzelnen Fällen beschrieben wurden, sind Chemotherapie, Gamma-Knife-Therapie und die Anwendung von Interleukinen [321] [321] [322] [324]. Rezidive sind mit ca. 40% häufig. Metastasierungen treten innerhalb der ersten 24 Monate auf [323] und finden sich in Lunge, Leber, Niere und Knochen [321]. Die Prognose ist insgesamt schlecht und zeigt eine Gesamtüberlebensrate von ca. 60% [106].
2.4.2.3.6 Biphänotypisches Sinunasalsarkom (BSS)
Das biphänotypische Sinunasalsarkom wurde in die WHO-Klassifikation der Tumore des Sinunasalsystems von 2017 als neue Entität aufgenommen. Begründet ist dies durch die definierende PAX3/MAML3-Translokation, welche es zu einer eigenen Entität macht. Bisher wurde das Malignom als Leiomyosarkom „fehlklassifiziert“ [325]. Das BSS wurde bisher nur im Sinunasaltrakt beschrieben.
Das BSS ist ein low-grade Spindelzellsarkom. Es tritt bevorzugt bei Frauen auf (w:m=3:1) und erstreckt sich über eine Altersspanne von 24–85 Jahren mit einem Altersdurchschnitt von 52 Jahren [106].
Dominierende Manifestationsorte sind Siebbeinzellen (57%) und Nasenhaupthöhle (54%), häufig mit grenzüberschreitendem Wachstum [326] und auch Infiltration der Orbita oder Fossa olfactoria.
Die Symptome sind unspezifisch. Patienten berichten von Nasenatmungsbehinderung, Druckgefühl und Gesichtsschmerzen. Makroskopisch zeigt sich eine polypoide, teils festere, rötlich und graue Tumormasse. Mikroskopisch ist die Zellularität mittelgradig mit fokalen zelldichteren Abschnitten im Tumor mit oberflächlich hamartomartiger Proliferation eingeschlossener ortsständiger Schleimhautdrüsen. Fokal zeigen sich vermehrte ektatische Gefäße mit Einblutungen und hämangioperizytomartigem Aspekt [326].
Das Malignom ist aufgrund der erst 2012 beschriebenen [327] und 2017 in die WHO-Klassifikation aufgenommen eigenständigen Entität weitestgehend unbekannt, dürfte aber häufiger als bisher angenommen sein, da viele Fälle bislang malignen peripheren Nervenscheiden- oder muskulären Tumoren zugeordnet wurden.
Langsames Wachstum und lokale Infiltration benachbarter Strukturen sind Charakteristika des BSS. Circa 50% der Patienten der ursprünglich beschriebenen Kohorte zeigten Lokalrezidive innerhalb eines Zeitraums von 9 Jahren. Von Fernmetastasierung wurde bisher nicht berichtet [327]. Auch existiert bislang nur ein dokumentierter Todesfall durch die Erkrankung [328].
Therapie der Wahl ist wie bei anderen Sarkomen die Tumorresektion mit ausreichendem Sicherheitsabstand. Adjuvante Radio- und Chemotherapie ist in Fällen mit fortgeschrittenem Tumorstadium einsetzbar. Die Effizienz der adjuvanten Radio- und Chemotherapie ist aufgrund der sehr geringen Fallzahlen unbekannt [329] [330].
2.4.2.3.7 Maligner peripherer Nervenscheidentumor/Neurofibrosarkom
Der maligne periphere Nervenscheidentumor (MPNST) entsteht an peripheren Nerven oder aus Transformationen gutartiger Tumore peripherer Nerven. In der Regel weist er eine Differenzierung zu einer der Komponenten der Nervenscheiden auf (z. B. Schwann-Zellen (malignes Schwannom), Fibroblasten oder perineurale Zellen).
Die Malignome treten vorwiegend bei erwachsenen Patienten mit einer großen Altersspanne und einem Altersgipfel innerhalb des 5. Lebensjahrzehnts auf. In ca. 20–25% der Fälle ist der maligne periphere Nervenscheidentumor mit einer Neurofibromatose Typ I assoziiert. In diesen Fällen sind die Patienten häufig jünger (zwischen 3. Und 4. Lebensjahrzehnt) [106] [331]. Eine Assoziation mit vorangegangener Radiotherapie wird diskutiert [332].
Die Inzidenz beträgt bei Patienten mit einer Neurofibromatose Typ I 1:3 500. Auf die gesamte Bevölkerung berechnet ist die Inzidenz 1:100 000 [333] [334]. Circa 20% aller malignen peripheren Nervenscheidentumore sind im Kopf-Hals-Bereich lokalisiert. Eine sinunasale Manifestation ist wesentlich seltener.
Manifestationsorte sind entlang von Hirnnerven, vorwiegend dem N. vestibularis und N. vagus lokalisiert [106] [331].
Klinische Symptome betroffener Patienten sind rasch progrediente, schmerzhafte Schwellungen und neurologische Ausfälle betroffener Nerven.
Maligne periphere Nervenscheidentumore sind unbekapselt und weisen ein höchst infiltratives Wachstum auf. Unterschiedlichste Zellmorphologien sind vorhanden, darunter spindelförmige, epitheloide und pleomorphe Zellen). Histologisch wird zwischen spindelzelligen und glandulären malignen peripheren Nervenscheidentumoren differenziert, die sich in der faszikelartigen Anordnung oder dem Vorhandensein von Becherzellen unterscheiden [106]. Die Malignome werden in Low- oder High-grade Entitäten entsprechend der Mitoserate, dem Vorhandensein atypischer Mitosen, Pleomorphismen und Nekrosen eingeteilt [332].
In der Regel sind diese Tumoren sehr aggressiv und haben eine schlechte Prognose. Die Tumorexzision mit ausreichendem Sicherheitsabstand ist Therapie der Wahl. Die Mehrzahl der malignen peripheren Nervenscheidentumore sind High-grade Sarkome, die zu Lokalrezidiven und Fernmetastasen neigen. 40–65% der betroffenen Patienten weisen Lokalrezidive auf, Fernmetastasierungen treten in 30–60% der Fälle auf, v. a. in Lunge, Leber, Gehirn, Knochen und Nebennieren. Regionale Lymphknotenmetastasierung ist selten, weshalb eine Neck dissection nicht standardmäßig durchgeführt werden sollte [333]. Eine adjuvante Radiotherapie, ggf. in Kombination mit einer Chemotherapie wirkt sich positiv auf das 5-Jahres Gesamtüberleben aus (65 vs. 38%) [335].
2.4.2.3.8 Sinunasales Synovialsarkom
Das Synovialsarkom ist ein mesenchymaler Tumor mit variabler epithelialer Differenzierung und enthaltenen Drüsenformationen. Typisch ist eine spezifische chromosomale Translokation (t(X;18)(p11;q11)), die zu einer Bildung eines SS18-SSX Fusionsgens führt [106] [336].
Das Synovialsarkom ist das häufigste Nicht-Rhabdomyosarkom-Weichteilsarkom bei Kindern, Heranwachsenden und jungen Erwachsenen mit einer Altersspanne zwischen dem 3. und 4. Lebensjahrzehnt.
Die Ätiologie ist stark assoziiert mit vorangegangener Radiotherapie [337] [337] [339]. Eine Manifestation im Sinunasaltrakt oder der Schädelbasis tritt äußerst selten auf.
Klinisch präsentiert sich eine palpable, meist tiefergelegene Raumforderung mit oder ohne Druckdolenz. Makroskopisch ist der Tumor gelb bis grau oder weißlich gestaltet. Langsam wachsende Synovialsarkome sind in der Regel gut abgegrenzt. Mikroskopisch könne monophasische (spindelzellförmig, kalzifizierend/ossifizierend, myxoid und schlecht differenziert) sowie biphasische Subtypen mit glandulären oder soliden Epithelzellen unterschieden werden. Schlecht differenzierte Tumore können Areale mit häufigen Mitosen und Nekrosen enthalten [106].
Die Therapie hat sich innerhalb der letzten Jahrzehnte nur wenig verändert und beinhaltet die Tumorresektion für umschriebene Befunde, häufig kombiniert mit einer Radio- oder Chemotherapie. Kombinierte Therapien werden je nach Stadium der Erkrankung angewandt. In einer großen Studie einer großen Studie kam bei Stadium I und II die Tumorresektion mit postoperativer Radiotherapie, bei Stadium III die Tumorresektion in Kombination mit einer Radiochemotherapie zum Einsatz [340]. Eine neoadjuvante Chemotherapie kann bei Patienten mit lokal fortgeschrittenem Tumorwachstum zum Einsatz kommen, bei denen eine mutilierende Operation erforderlich wäre. Erste Ergebnisse der Immuntherapie, die eine Aktivität bei NY-ESO-1, Trabectedin und einer Vielzahl von Angiogenese-Inhibitoren zeigen, sind vielversprechend [341].
2.4.2.4 Borderline- und niedrig maligne Entitäten des Weichgewebes
2.4.2.4.1 Aggressive Fibromatose vom Desmoid-Typ
Die aggressive Fibromatose ist eine klonale, spindelzellige Neoplasie mit infiltrativem Wachstum. Fernmetastasen treten nicht auf. Die Begriffe Desmoid-Fibromatose und aggressive Fibromatose werden synonym mit der aggressiven Fibromatose vom Desmoid-Typ verwendet.
Die Inzidenz wird auf 1:250 000 bis 1:500 000 geschätzt. Das Erkrankungsalter reicht von 15–60 Jahren, ca. 30% der Fälle treten im Kindesalter auf. Die Manifestation im Kopf-Hals-Bereich betrifft ca. 15% aller aggressiven Fibromatosen. Die sinunasale Manifestation ist noch wesentlich seltener [106] [342] [343].
Die Ätiologie ist ungeklärt. Eine Assoziation mit dem Gardner-Syndrom (siehe 2.4.1.2.1) und der familiären kolorektalen Polyposis besteht [344] [345].
Aggressive Fibromatosen vom Desmoidtyp zeigen schlecht abgrenzbare Läsionen mit fokal infiltrativem Wachstum, die sich makroskopisch als feste, weißliche Läsionen mit trabekulärem Schnittmuster präsentieren. Immunhistochemisch zeigen Desmoide eine nukleäre Anfärbung mit β-Catenin und häufig zusätzlichem zytoplasmatischem Hintergrund [326]. Faszikuläres Wachstum mit spindelförmigen Zellen und milden Kernpleomorphismen können visualisiert werden. Atypische Mitosen und Nekrosen sind nicht vorhanden. Das Stroma kann variabel kollagenisiert und myxoid oder mukös anmutend konfiguriert sein.
Eine adäquate Schmerztherapie ist für Patienten mit aggressiver Fibromatose erforderlich. Emori et al. konnten in einer Sammlung von 16 Fällen zeigen, dass der Desmoid-Tumor-assoziierte Schmerz mit einer Überexpression an Cyclooxygenase 2 assoziiert ist [346].
Grundsätzlich ist die vollständige chirurgische Resektion Therapie der Wahl. Aufgrund der anatomischen Nähe zu kritischen Strukturen kann bei Lokalisation im Sinunasaltrakt eine systemische medikamentöse Therapie (Anti-Östrogen-Therapie, nichtsteroidale Antiphlogistika), Chemotherapie (Vinblastin/Vinorelbine, pegyliertes liposomales Doxorubicin), Tyrosin-Kinase.Inhibitoren (z. B. Imatinib, Sorafenib) oder eine Radiotherapie zurückgegriffen werden [326] [343] [347]. Innerhalb der letzten Jahre wurde allerdings zunehmend eine „wait and scan“ Strategie propagiert, da verfügbare Daten zeigten, dass nur ein geringer Anteil von aggressiven Fibromatosen eine Progression aufweist [348] und diese meist innerhalb der ersten 36 Monate nach Diagnosestellung [349].
Die Prognose ist gut bei R0-Resektionen. Bei R1-Resektionen kommt es in der Regel innerhalb von<2 Jahren zu einem Rezidiv [350].
2.4.2.4.2 Sinunasales Glomangioperizytom
Das Sinunasale Glomangioperizytom ist eine Spindelzellneoplasie, die in der WHO-Klassifikation der Kopf-Hals-Tumore als neue Entität aufgenommen wurde. Zuvor wurde die Entität als sinunasales Hämangioperizytom bezeichnet. Innerhalb der letzten 60 Jahre war der Begriff Hämangioperizytom gebräuchlich, um eine Vielzahl von Neoplasien zu beschreiben, die ähnliche morphologische Eigenschaften aufwiesen. Diese Eigenschaften betrafen ca. 15% aller Weichgewebs-Neoplasien [351], was zu Konfusion bei der Etablierung eines spezifischen Therapieregimes führte [352]. Heute bezeichnet der Terminus Hämangioperizytom keine eigene Neoplasie sondern eher ein Wachstumsmuster, das von mehreren Neoplasien geteilt wird, die allerdings sehr unterschiedlich sind [353].
Glomangioperizytome repräsentieren ca. 0,5% aller sinunasalen Neoplasien. Bisher sind etwas mehr als 100 Fälle beschrieben [352]. Der Altersgipfel liegt im 7. Lebensjahrzehnt mit einer leichten Prädilektion bei weiblichen Patienten [106].
Häufigster Manifestationsort ist die Nasenhaupthöhle mit Ausdehnung in die angrenzenden Nasennebenhöhlen. Isolierte Fälle in den Nasennebenhöhlen sind selten. Das Glomangioperizytom tritt meist unilateral auf.
Betroffen Patienten berichten von Nasenatmungsbehinderung mit Druckgefühl und Epistaxis. Die Beschwerdedauer bis zur Diagnose beträgt häufig mehr als ein Jahr [354].
Makroskopisch zeigt der Tumor ein polypoides, kräftig rotes bis pinkes Bild. Die Oberfläche ist weich und fleischig. Durchschnittlich beträgt die Tumorgröße bei Diagnosestellung 3 cm. Mikroskopisch zeigt sich ein unbekapseltes Wachstum unter einer intakten Epithelschicht mit nur selten Erosionen v. a. bei größeren Tumoren. Charakteristisch ist ein musterloser, diffuser Aufbau mit partiell faszikulärer Zellanordnung, getrennt von Gefäßplexus aus Kapillaren bis hin zu großen Kavernen und eine prominente, azelluläre Hyalinisierung [106]. Zellatypien fehlen. Β-Catenin ist im Gegensatz zu den Desmoiden (siehe 2.4.2.4.1) in fast allen Tumorzellen kräftig nachweisbar [326].
Glomangioperizytome wachsen langsam und weisen eine sehr gute Überlebensrate auf. Rezidive treten in bis zu 40% der Fälle auf und sind meist Folge einer unzureichenden Resektion. Invasives Wachstumsverhalten tritt in der Regel ab einer Tumorgröße über 5 cm auf [354] [355].
Die Tumorresektion gilt trotz der geringen Fallzahlen als Therapiestandard. Radio- und Chemotherapie kann bei unresektablen Tumoren oder Fernmetastasierung zum Einsatz kommen. Adjuvante Radiotherapie ist möglich, um die lokale Tumorkontrolle zu verbessern. Je nach Ausprägung der Vaskularisation ist eine präoperative Embolisation empfehlenswert, um den intraoperativen Blutverlust zu reduzieren [356] [356] [358].
2.4.2.4.3 Sinunasaler solitärer fibröser Tumor
Der solitäre fibröse Tumor basiert auf einer Fusion der Gene NAB2 und STAT6 und weist einen fibroblastischen Phänotyp mit sich stark verzweigenden Gefäßstrukturen auf.
Synonym wird der Begriff Hämangioperizytom oder Riesenzell-Angiofibrom verwendet.
Der solitäre fibröse Tumor ist eine Rarität und ist für weniger als 0,1% aller sinunasalen Neoplasien verantwortlich. Vor allem Erwachsene sind betroffen ohne eindeutige Präferenz für ein Geschlecht [106].
Der Tumor tritt hauptsächlich in der Nasenhaupthöhle auf und führt bei betroffenen Patienten zu Nasenatmungsbehinderung und Epistaxis sowie unspezifischen Beschwerden wie Druckgefühl.
Makroskopisch zeigt sich ein polypoider, fest strukturierter, weißlicher Tumor, der aufgrund des vorgegebenen engen Raums im Sinunasaltrakt üblicherweise klein ist [359] [360]. Histologisch zeigen sich submukös liegende, pseudobekapselte Tumore mit variablen Zellformationen, darunter spindelförmige Zellformationen, die wahllos angeordnet erscheinen. Die Gefäße sind sternförmig bis hirschgeweihartig angeordnet [106] [325].
Therapie der Wahl ist die vollständige Tumorresektion, welche üblicherweise zu einer kurativen Situation führt. Patientenalter über 55 Jahre, Tumorgröße über 15 cm, nekrotische Tumorareale und mehr als 4 Mitosen pro 10 hochauflösenden Gesichtsfeldern legen aggressiveres Wachstum nahe [361] [362]. Eine adjuvante Radiotherapie scheint in diesen Fällen v. a. bei unvollständiger Resektion und Lokalrezidiven als zusätzliche Behandlung möglich zu sein [363]. Die Effizienz dieser Therapie ist allerdings aufgrund der extrem niedrigen Fallzahlen nicht abschließend zu bewerten.
2.4.2.4.4 Epitheloides Hämangioendotheliom
Das epitheloide Hämangioendotheliom ist eine low- bis intermediate-grade Neoplasie aus Zellen, die einen endothelialen Phänotyp, epitheloide Morphologie und ein hyalinisiertes, chondroides oder basophiles Stroma aufweisen [106].
Betroffen sind v. a. Erwachsene. Aufgrund des extrem seltenen Auftretens und der schwierigen Differenzialdiagnostik ist eine Inzidenz-Abschätzung nur schwer möglich.
Das Vorkommen im Kopf-Hals-Bereich ist sehr selten. Der Entstehungsort liegt meist im Weichgewebe, der Haut oder dem Knochen. Extrem selten können Lymphknoten als primäre Manifestationsorte vorkommen [364] [364] [365] [367].
Epitheloide Hämangioendotheliome wachsen sehr langsam, infiltrieren umgebende Strukturen und metastasieren selten [368]. Die Symptome sind meist unspezifisch. Epistaxis kann je nach Lokalisation auftreten.
Makroskopisch zeigt sich eine noduläre Tumormasse mit blasser, teils rötlicher und teils hämorrhagischer Schnittfläche. Histologisch zeigen sich endotheliale und histiozytäre Zellen, die in kurzen, fadenartigen Formationen in einem myxohyalinene Stroma angeordnet sind. Die mitotische Aktivität ist niedrig. Multizelluläre vaskuläre Kanäle können vorhanden sein. Endotheliale Marker sind nachweisbar (CD31, ERG, FLI1). In den meisten Fällen ist eine WWTR1-CAMTA1 Genfusion vorhanden [106].
Die meisten Fälle verlaufen indolent. Fallberichte mit tumorbedingter Mortalität existieren [369] [369] [371].
Die Therapie der Wahl ist die radikale Tumorresektion [372], die in bis zu 85% zu Rezidivfreiheit führt [373]. Eine medikamentöse Therapie kann kurativ oder adjuvant durchgeführt werden. Sie beinhaltet eine Kombination aus Corticosteroiden, zytotoxische Wirkstoffe, Thrombozytenaggregationshemmer und Antifibrinolytika und Interferon-alpha. Für Fälle, in denen eine R0-Resektion aufgrund hoher Morbidität nicht erreichbar ist, kann zudem eine Radiotherapie diskutiert werden [373] [374].
2.4.2.5 Hämatolymphoide Tumore
2.4.2.5.1 Extranodales NK/T-Zell Lymphom
Das extranodale NK/T-Zell Lymphom ist ein extranodales Lymphom mit zytotoxischem Phänotyp und einer zwingenden Assoziation mit dem Epstein-Barr-Virus. Synonym werden die Begriffe angiozentrisches Lymphom und letales Mittellinien-Granulom verwendet.
Das Malignom hat eine höhere Prävalenz in Südostasien und der indigenen Bevölkerung von Mexiko und Zentral- sowie Südamerika. Hier repräsentiert es bis zu 10% der Non-Hodgkin Lymphome. Im Gegensatz hierzu ist dieser Anteil in Nordamerika und Europa unter 1%. Die Prävalenz der Erkrankung wird in Europa auf unter 9:1 000 000 Einwohner geschätzt [375].
Das extranodale NK/T-Zell Lymphom wächst destruierend im oberen Aerodigestivtrakt und manifestiert sich in der Nasenhaupthöhle, den Nasennebenhöhlen und entlang des Waldeyerschen Rachenrings [106].
Erste Symptome sind Nasenatmungsbehinderung und Epistaxis. Das infiltrative Wachstum führt häufig zu Perforationen des Nasenseptums oder des harten Gaumens und zu Einwachsen in die Haut, an der bei Durchbruch ulzerierende Läsionen entstehen. Bei Lokalisation innerhalb der Nasennebenhöhlen können Symptome einer chronischen Rhinosinusitis die eigentliche Erkrankung kaschieren [106]. Funktionseinschränkungen stellen sich bei okulärer oder zerebraler Infiltration ein.
Histopathologisch zeigt sich ein diffus in das Gewebe infiltrierender Tumor mit angiozentrischem bzw. angioinvasivem Wachstumsmuster und großen Tumornekrose-Arealen. Die neoplastischen Zellen variieren in Größe und Anzahl der irregulär geformten Zellkerne. Immunhistochemisch exprimiert der Tumor CD3, zytotoxische Marker und CD56 [376] [377].
Aufgrund der strikten EBV-Assoziation und der klaren ethnischen Prädisposition wird ein Gendefekt der Immunantwort betroffener Patienten gegenüber einer EBV-Infektion vermutet [378] [379].
Die Prognose ist im Vergleich zu anderen T-Zell Lymphomen schlecht mit einer medianen Überlebensrate von 7,8 Monaten und einer 5-Jahres Überlebensrate von 40% [15]. Die plasmatische EBV-DNA Last ist von diagnostischer und prognostischer Signifikanz. Sie wurde zusammen mit den PET CT Ergebnissen in die prognostischen Algorithmen integriert [106] [380]. Aktuelle Chemotherapieregimes erreichen langfristige Remissionen in 70–80% der Fälle in Stadium I/II und ca. 50% in Stadium III/IV [381] [382].
2.4.2.5.2 Extraossäres Plasmozytom
Das extraossäre Plasmozytom stellt eine raumfordernde Proliferation monoklonaler Plasmazellen mit extraossärer Manifestation ohne das zugrundeliegende multiple Myelom dar. Wichtig ist die Unterscheidung von B-Zell Lymphomen mit plasmozytischer/plasmablastischer Differenzierung, speziell vom MALT Lymphom und dem plasmoblastischen Lymphom [106].
Das mediane Patientenalter bei Diagnosestellung beträgt ca. 60 Jahre und es besteht eine erhöhte Prävalenz bei Patienten männlichen Geschlechts (m:w=3–4:1) [106] [383] [384].
Ca. 80% der extraossären Plasmozytome manifestieren sich innerhalb der oberen Atemwege, vorwiegend der Nasenhaupthöhle und den Nasennebenhöhlen. Die weltweite Inzidenz wird auf 0,021 bis 4:100 000 Einwohner geschätzt [385].
Klinisch zeigen sie solitäre Raumforderungen, die zu Atemwegseinengungen, Epistaxis, Gesichtsschmerzen, chronisch sinunasalen Beschwerden mit Rhinorrhoe und je nach Infiltration zu Hirnnervenausfällen und einer Protrusio bulbi führen können.
Unter 25% der Patienten weisen ein monoklonales Serum-Paraprotein auf, typischerweise vom IgA-Typ. Diagnostische Merkmale des multiplen Myeloms fehlen [386] [387].
Histologisch kann eine diffuse Infiltration gut, mittelgradig oder nur gering differenzierter Plasmazellen und gelegentlich Amyloidablagerungen festgestellt werden. Mittelgradig und gut differenzierte extraossäre Plasmozytome müssen von B-Zell Lymphomen, insbesondere vom MALT Lymphom mit extensiver plasmozytischer Differenzierung abgegrenzt werden. Gering differenzierte extraossäre Plasmozytome sind von plasmoblastischen Lymphomen zu differenzieren [388] [389]. Die Zellen exprimieren häufig Merkmaler einer plasmozytischen Differenzierung (C138, C38, VS38, MUM1/IRF4) [390]. Monotypische Immunglobulin Leichtketten können typischerweise festgestellt werden.
Therapie der Wahl ist die lokale Radiotherapie, welche eine wesentlich bessere Prognose als bei multiplem Myelom erbringt. Die krankheitsspezifische 5- und 10-Jahres Überlebensrate wird in der aktuellen Literatur mit bis zu 82 bzw. 76% angegeben [391] [391] [393]. Lokalrezidive oder Versprengung in andere extraossäre Lokalisationen sind möglich. Circa 15% der Patienten entwickeln im Verlauf ein multiples Myelom [383].
2.4.2.5.3 Langerhans-Zell-Histiozytose
Im Rahmen der Langerhans-Zell-Histiozytose (LZH) kommt es zu einer Proliferation und Akkumulation von Langerhans-Zellen in unterschiedlichen Gewebearten. Die Prävalenz in der europäischen Bevölkerung wird auf 1–2:100 000 geschätzt. Eine primäre Manifestation der LZH in der Nase, den Nasennebenhöhlen oder an der Schädelbasis ist noch deutlich seltener anzutreffen. Genaue Zahlen hierzu existieren nicht. Besonders häufig ist die Erkrankung bei Kindern zu finden. Der Altersgipfel betroffener Patienten liegt zwischen 1 und 4 Jahren [394] [395]. Typische Manifestationen der LZH finden sich innerhalb des Knochens (80%), in der Haut (35%) sowie an der Hypophyse (25%). Seltener betroffene Organsysteme sind Lunge, Leber und das hämatopoetische System (15–20%) [396]. Schwere Verläufe können zu einer sklerosierenden Cholangitis oder neurodegenerativen Veränderungen in bis zu 2% der Fälle führen.
Bei Befall der Nase oder der Nasennebenhöhlen stellen sich zumeist Druckgefühl und Schmerzen über der befallenen Region ein. Bei Befall des Sinus frontalis oder maxillaris kann es zu Schwellungen im Gesichtsbereich kommen [397]. Aufgrund der raumfordernden Wirkung der Manifestation der LZH sind Affektionen benachbarter Strukturen wie z. B. dem Nervus opticus möglich [398].
Bildmorphologisch präsentiert sich die Erkrankung mit gut abgrenzbaren, fast „ausgestanzt“ wirkenden knöchernen Läsionen und Beteiligung des angrenzenden Weichgewebes. Läsionen ohne knöcherne Erosionen wurden ebenfalls demonstriert [399] [400]. In einer Untersuchung von 163 Patienten mit LZH konnte in 55% der Fälle eine magnetresonanztomografische Beteiligung der Nasennebenhöhlen oder der Mastoide diagnostiziert werden [401].
Aktuelle Studien haben gezeigt, dass bei Patienten mit disseminierter Erkrankung eine BRAF V600E Mutation nachgewiesen werden kann, die ebenfalls in malignen Melanomen auftritt [402] [402] [404]. Die Möglichkeit einer Anwendung einer „Targeted Therapy“ mit Inhibitoren, die bereits im Rahmen eines Malignen Melanoms verwendet werden, wird derzeit untersucht. Nicht zuletzt aufgrund dieser Mutation bleibt die Debatte, ob es sich bei der LZH um eine maligne Erkrankung mit variierender klinischer Manifestation handelt, bestehen.
Aufgrund der nicht gänzlich geklärten Pathogenese der LZH ist die Therapie empirisch und hängt von der jeweiligen Manifestation und dem Grad des systemischen Befalls ab. Die Diagnose wird über eine Gewebeprobe gesichert. Die Exzision oder Kürettage befallener Areale kann eine effektive Therapie für unifokale Manifestationen sein [405]. Die Prognose ist bei unifokaler Manifestation gut. Die Gabe von 125 mg Methylprednisolon wird aufgrund der inhibitorischen Wirkung auf die Osteolyse empfohlen. Radiotherapie wird vorwiegend bei Rezidiven eingesetzt. Bei systemischem Befall mit multifokaler Ausbreitung ist die Prognose deutlich schlechter. Therapeutisch werden medikamentöse Therapien mit Vinblastin, Prednison, Etoposid und Methotrexat in verschiedenen Kombinationen verwendet [406].
2.4.2.6 Neuroektodermale und melanozytäre Tumore
2.4.2.6.1 Ewing-Sarkom, primitiver neuroektodermaler Tumor
Das Ewing Sarkom und der primitive neuroektodermale Tumor sind primitive, rund-/kleinzellige high-grade Sarkome mit variabler neuroektodermaler Differenzierung. Charakteristisch ist eine Translokation zwischen dem EWSR1 Gen und Chromosom 22 sowie einem Mitglied der ETS-Transkriptionsfamilie [106].
Synonym werden die Begriffe peripherer neuroektodermaler Tumor, peripheres Neuroepitheliom, peripheres Neuroblastom oder adultes Neuroblastom verwendet.
Ewing Sarkome und primitive neuroektodermale Tumore manifestieren sich nur in 2–10% der Fälle im Kopf-Hals-Bereich und weisen eine höhere Inzidenz beim männlichen Geschlecht auf. Vorwiegend sind Kinder und junge Erwachsene betroffen [106].
Manifestationen im Kopf-Hals-Bereich sind Schädel- und Kieferknochen, deutlich weniger sind die Nasennebenhöhlen oder die Nasenhaupthöhle betroffen. Häufigster Manifestationsort des Sinunasaltrakts ist der Sinus maxillaris und die Nasenhaupthöhle. Infiltrationen in die Orbita oder nach intrakraniell sind möglich [407] [408]. [Abb. 15] zeigt die magnetresonanztomografische Darstellung des Schädels eines 9-jährigen Patienten, der durch eine Abduzensparese linksseitig aufgefallen war.

Schmerzen, progrediente Schwellungen, Nasenatmungsbehinderung mit raschem Progress sind typische Symptome betroffener Patienten.
Makroskopisch zeigen sich polypoide oder multilobuläre, grau/weißliche, teils hämorrhagische Tumore mit Ulcerationen. Histologisch ist ein ausgesprochen zellulärer Tumor mit diffusem bis lobulärem Wachstum und trabekulärem oder strangartigem Aufbau festzustellen. Einheitliche kleine Zellen mit runden bis ovalen Zellkernen, diskretes nukleäres Chromatin, blasses Zytoplasma und undeutliche Zellgrenzen sind weitere Merkmale. Die mitotische Aktivität ist mit 5–10 Mitosen pro hochauflösenden 10 Gesichtsfeldern variabel ausgeprägt [106]. In 90–95% der Fälle existiert eine konsistente, reziproke Translokation zwischen den EWSR1 Gen auf Chromosom 22 und dem FLI1 Gen auf Chromosom 11 [409].
Neben der MRT und CT ist die PET-CT zum Staging empfohlen, um Fernmetastasierung auszuschließen bzw. korrekt zu lokalisieren und das eventuell erforderliche chirurgische Vorgehen besser planen zu können [410]. Sofern möglich, ist eine Exzision des Befundes mit ausreichendem Sicherheitsabstand anzustreben. Obwohl Ewing Sarkome als strahlensensitiv eingestuft werden, ist die Anzahl von Patienten, die innerhalb der letzten 30 Jahre primär radiotherapiert wurden, stetig gesunken. Ein Grund hierfür ist die Spättoxizität und das Risiko von sekundären Malignomen nach Radiotherapie, da besonders Kinder von der Erkrankung betroffen sind. Die Modifikation von Chemotherapien hat innerhalb der letzten Jahrzehnte die Überlebensraten von ursprünglich ca. 10% deutlich erhöht. Die derzeitige 5-Jahres Überlebensrate für sinunasale Ewing-Sarkome/primitive neuroektodermale Tumore ohne Metastasierung liegt bei 50–75% und ist damit besser als für andere Lokalisationen [408] [411]. Chemotherapie-Schemata sind VACA (Vincristin, Doxorubicin, Cyclophosphamid, Actinomycin), VAIA (Ifosfamid anstelle von Cyclophosphamid) oder EVAIA (zusätzlich Etoposid) und VIDE (ohne Actinomycin). Trotz der guten Prognoseentwicklung ist das Outcome von Patienten mit Metastasen deutlich schlechter [412].
2.4.2.6.2 Olfaktoriusneuroblastom
Olfaktoriusneuroblastome sind maligne neuroektodermale Neoplasien. Entsprechend ihrer Genese sind sie häufig in den kranialen Anteilen der Nasenhaupthöhle entlang der Regio olfactoria lokalisiert. Synonym wird der Begriff Aesthesioneuroblastom verwendet.
Die Inzidenz wird mit ca. 4:10 000 000 angegeben. Insgesamt repräsentiert die Entität ca. 3% aller Nasennebenhöhlenmalignome [106] [413]. Das Alter betroffener Patienten reicht in den in der Literatur beschriebenen Fällen von 2 bis 90 Jahren mit einem Altersgipfel zwischen dem 5. und 6. Lebensjahrzehnt. Männer sind häufiger betroffen (1,2:1). Eine ethnische oder familiäre Prädilektion existiert nicht.
Der Tumor kann sich entlang der Lamina cribrosa, dem medialen Anteil der mittleren Nasenmuschel oder dem kranialen Anteil des Nasenseptums manifestieren. Ursprungsorte sind das vomeronasale Organ, das Ganglion sphenopalatinum, entsprechend der Embryologie die olfaktorische Plakode oder der Nervus terminalis, der die Ethmoidspalte des anterioren Anteils der Lamina cribrosa ausfüllt [414]. Ektope Manifestationen im Siebbein sind möglich, allerdings extrem selten für alle anderen Sinus [415] [416]. Aufgrund des Entstehungsortes ist die Schädelbasis grundsätzlich in das Tumorgeschehen involviert.
Initial weisen Olfaktoriusneuroblastome häufig Beschwerden auf, die denen gutartiger Läsionen ähneln, weshalb es häufig zu einer späten Diagnosestellung kommt. Nasenatmungsbehinderung und gelegentlich Epistaxis werden von den Patienten berichtet. Cephalgien, Rhinorrhoe, Epiphora und Visusstörungen sind Symptome eines bereits ausgedehnteren Tumorwachstums. Die Anosmie ist trotz der Lokalisation an der Lamina cribrosa mit<5% selten. Paraneoplastische Syndrome treten in ca. 2% der Fälle auf.
Klinisch präsentiert sich eine Tumormasse, die von der Lamina cribrosa ausgeht und sich über weite Teile dieser erstreckt. Meist ist das Tumorwachstum unilateral und imponiert polypoid, weich mit rot-grauer Oberfläche bei intakter Mucosa. Computer- und vor allem Magnetresonanztomografie ermöglichen die Ausdehnungsbestimmung ([Abb. 16]) und Darstellung einer Infiltration der Orbita, was beispielhaft an [Abb. 17] zu erkennen ist.


Es existieren verschiedene Staging-Systeme (Kadish [417], Morita [418] und nach TNM [419]), wobei die Einteilung nach Kadish die weiteste Verbreitung zeigt.
Olfaktoriusneuroblastome werden in low grade- und high grade-Entitäten unterteilt. Low grade Olfaktoriusneuroblastome zeigen scharf demarkierte, submukös wachsende Zellnester, die oft durch vaskuläres oder hyalinisiertes Bindegewebe voneinander getrennt sind. Sogenannte Homer Wright Rosetten/Pseudorosetten, in denen sich neoplastische Zellen palisadenartig um die zentral liegende neurale Matrix verteilen, sind charakteristisch. High grade Olfaktoriusneuroblastome zeigen Tumornekroseareale, Pleomorphismen, eine gesteigerte Mitoserate sowie ein weniger offensichtliches lobuläres Wachstum [106].
Das bekannteste Grading-System stammt von Hyams et al. [420]. In diesem wird je nach Differenzierungsgrad zwischen Grad I (hochdifferenziert) und Grad IV (niedrig differenziert) unterschieden. Kriterien sind Tumorarchitektur, mitotische Aktivität, nukleäre Polymorphismen, fibrilläre Matrizen und Rosetten, Nekroseareale, glanduläre Proliferation und Kalzifikationen.
In einer 2009 publizierten Arbeit von Mao et al. konnte gezeigt werden, dass der PTCH1, GLI1 und GLI2 Signalweg in die Pathogenese der Olfaktoriusneuroblastome involviert sind, was eine zentrale Rolle des Sonic Hedgehog Signalwegs nahelegt [421].
Primäre Therapieoption ist die Tumorresektion, gefolgt von einer lokalen Radiotherapie. Entgegen der traditionellen, 1963 erstmals beschriebenen kraniofazialen Resektion [422] wird heute die endoskopische Resektion des Tumors favorisiert. Entgegen der ursprünglichen Annahme, dass nur die en-bloc-Resektion des Tumors langfristige Rezidiv-Freiheit gewähren könnte, zeigen endoskopische Verfahren ein ähnlich gutes onkologisches Outcome bei allerdings deutlich weniger invasiven Eingriffen [423]. Fortgeschrittenes Tumorwachstum kann zusätzlich eine Chemotherapie erforderlich machen. High grade Tumore nach Hyams sprechen hierauf besser an, weshalb in Einzelfällen auch eine Induktions-Chemotherapie diskutiert werden kann [424].
Eine Meta-Analyse von Dulguerov et al analysierte die Krankheitsverläufe in Abhängigkeit der Therapieschemata und zeigte das beste Outcome für Patienten, die eine Tumorresektion gefolgt von einer lokalen Radiotherapie erhalten hatten. Patienten dieser Gruppe zeigten eine 2-Jahres Überlebensrate von 65%, verglichen mit 48 und 37% bei alleiniger Tumorresektion bzw. alleiniger Radiotherapie [425]. Aktuelle Studienergebnisse zeigen zudem ein signifikant verbessertes krankheitsspezifisches Überleben für Patienten im Stadium T3/T4, bei denen eine Tumorresektion und Strahlentherapie durchgeführt wurde, im Vergleich zu einer alleinigen chirurgischen Therapie [426].
2.4.2.6.3 Maligne Schleimhautmelanome
Schleimhautmelanome sind maligne Neoplasien, die aus Melanozyten der Schleimhaut entstehen und die sich biologisch von kutanen Melanomen unterscheiden. Die Ätiologie ist unbekannt.
Schleimhautmelanome sind für weniger als 1% aller malignen Melanome verantwortlich [427] und repräsentieren ca. 4% aller sinunasalen Tumore. Die Altersspanne ist weit gefächert und weist einen Altersgipfel innerhalb des 7. Lebensjahrzehnts auf [106].
Häufigster Manifestationsort ist die Nasenhaupthöhle mit dem Nasenseptum und deutlich seltener der Nasopharynx oder Sinus maxillaris [428] [429]. Aufgrund der Abschirmung von Sonneneinstrahlung scheint das sinunasale Schleimhautmelanom eine unterschiedliche Biologie gegenüber dem kutanen malignen Melanom zu besitzen. Etwa 20% der betroffenen Patienten weisen bei Diagnosestellung multifokale Herde auf, etwa 40% der diagnostizierten Melanome sind amelanotisch [430].
Maligne Schleimhautmelanome können sich als plane, pigmentierte Schleimhautveränderungen ([Abb. 18]) oder als polypöse, raumfordernde Tumorformationen präsentieren ([Abb. 19] und [ 20]). Symptome bei der Erstvorstellung sind häufig Epistaxis und Obstruktion der Nase bzw. Nasenatmungsbehinderung. Derart unspezifische Beschwerden führen häufig zu einer Diagnosestellung in höheren Tumorstadien.



Makroskopisch weist das Schleimhautmelanom eine polypoide Formation auf. Es kann pigmentiert und brüchig bis hellbraun oder grau und fest konfiguriert sein [106]. Mikroskopisch zeigen sich häufig Ulzerationen der Schleimhautbedeckung mit variabler Zellmorphologie, die von epitheloiden/undifferenzierten Zellen zu spindelformigen, plasmazytoiden und rhabdoiden Zellen mit teilweisen prominenten Kernen reicht. Atypische Mitosen sind häufig zu beobachten. Knapp die Hälfte der Malignome können eine amelanotische Ausprägung zeigen, was die Anzahl der möglichen Differenzialdiagnosen deutlich erhöht (u. a. Olfaktoriusneuroblastom, Rhabdomyosarkom, Sinunasales undifferenziertes Karzinom, schlecht differenziertes Plattenepithel-Karzinom) [106].
Im Gegensatz zu kutanen Melanomen sowie Aderhautmelanomen zeigen sich höhere Raten von KIT- und NRAS-Mutationen. BRAF Mutationen hingegen sind selten [431] [432].
Entscheidend für Staging und Prognose ist die korrekte Differenzierung eines mukosalen Melanoms von einer sinunasalen Metastasierung eines kutanen malignen Melanoms. Fernmetastasierung und fortgeschrittenes Patientenalter sind die entscheidendsten Prognosefaktoren. Entsprechend der 7. Ausgabe des „Cancer Stanging Manuals des American Joint Committee on Cancer“ besteht im Stadium T3 und T4 eine 5-Jahres Überlebensrate von unter 30% [433] [433] [435].
Gegenwärtige Behandlungsempfehlungen befürworten die weiträumige lokale Exzision, sofern möglich [436]. Die Rolle der postoperativen Radiotherapie ist umstritten. Eine Meta-Analyse aus dem Jahr 2012 konnte keine höhere Überlebensrate feststellen [437]. Andere Studienergebnisse belegen eine gesteigerte 3-Jahres Überlebensrate von 18 auf 30% sowie eine reduzierte lokale Rezidivrate nach postoperativer Radiotherapie [438] [439]. Anti-PD-1 Antikörper sowie Anti-CTLA-4 Antikörper Therapien zeigen vielversprechende Ergebnisse bei fortgeschrittenen oder metastasierten Schleimhautmelanomen, allerdings ist die Evidenz zu gering, um die klinische Anwendung zu rechtfertigen [436] [440].
2.4.2.7 Keimzelltumore
2.4.2.7.1 Sinunasales Teratokarzinosarkom
Teratokarzinosarkome sind maligne Neoplasien mit histologischen Eigenschaften sowohl von Teratomen als auch Karzinosarkomen ohne bösartige Keimzellbestandteile. Die Begriffe Malignes Teratom, Blastom, Teratokarzinom und teratoides Karzinosarkom werden synonym verwendet.
Teratokarzinosarkome sind sehr seltene Tumore, die hauptsächlich bei männlichen Erwachsenen auftreten (mittleres Patientenalter 60 Jahre).
Am häufigsten tritt der Tumor in der Nasenhaupthöhle, gefolgt von Sinus ethmoidalis und maxillaris auf. In etwa 20% der Fälle kann eine intrakranielle Beteiligung festgestellt werden [441].
Die Diagnose stellt sich anhand des Nachweises von malignen epithelialen Elementen und 2 oder mehr malignen mesenchymalen Komponenten wie z. B. Fibroblasten, Knorpel, Knochen oder glatter Muskulatur. Diese Kombinationen können primitiven bronchialen oder intestinalen Strukturen ähneln, die im Sinonasaltrakt fremd wirken [442]. Die teratoiden Elemente sind wesentliche diagnostische Faktoren des sinunasalen Teratokarzinosarkoms. Fetal anmutendes, klarzelliges Plattenepithel und tubuläre oder glanduläre Formationen sind weitere Kriterien für die Diagnosestellung. Stammzell-Anteile sind beim Teratokarzinosarkom nicht vorhanden [443] [443] [444] [445] [447].
Die Behandlung des sinunasalen Teratokarzinosarkoms ist aufgrund der hohen Malignitätsrate und der schlechten Prognose schwierig. Um realistische Chancen auf ein längerfristiges tumorfreies Überleben zu haben, wird eine radikale Tumorresektion gefolgt von einer Radiotherapie empfohlen [448]. In Fällen einer intrakraniellen Ausdehnung wurden kombinierte intra- und extrakranielle Herangehensweisen beschrieben, um en-bloc Resektionen zu realisieren [449]. Eine Übersichtsarbeit von Misra et al. identifizierte 5 Berichte, in denen die Tumorresektion endoskopisch unterstützt wurde [441]. Die Rolle einer adjuvanten Chemotherapie ist aufgrund nur weniger Fallberichte nicht gesichert. Aufgrund der extremen Seltenheit sind die Charakteristika und optimale Therapieschemata nicht definiert.
Lymphknoten- und Fernmetastasierungen sind häufig. Die berichteten Überlebensraten variieren von 50–70% in unterschiedlichen Analysen [106] [441].
Zusammenfassung
Die vorliegende Übersicht stellt seltene Erkrankungen der Nase, der Nasennebenhöhlen sowie der vorderen Schädelbasis mit Fokus auf Fehlbildungen, Belüftungs- und Funktionsstörungen wie auch Tumorerkrankungen vor. Ein Anspruch auf Vollständigkeit kann aufgrund der Komplexität des anatomischen Bereichs und der Vielzahl möglicher Erkrankungen (Funktionsstörungen, Fehlbildungen, Tumore) nicht erhoben werden.
In vielen Fällen ist die adäquate Therapie einer seltenen Erkrankung komplex und bedarf umfangreicher Recherche der in der Literatur beschriebenen Erfahrungen. Vor allem bei seltenen benignen und malignen Neoplasien ist der Nutzen einer adjuvanten Therapie oftmals nicht geklärt und erfordert Erfahrung, die allerdings aufgrund der geringen Fallzahlen in vielen Zentren nicht vorgehalten werden kann. Auch muss innerhalb der Gruppe der seltenen Erkrankungen eine Abstufung vorgenommen werden, da extreme Unterschiede in den Inzidenzen der einzelnen Krankheitsbilder vorliegen. Eine Inzidenz von weniger als 5:10 000 erfüllt zwar die Definition einer seltenen Erkrankung, führt aber an einem großen Zentrum für Hals-, Nasen-, Ohrenheilkunde, Kopf- und Halschirurgie zu einer durchaus nennenswerten Anzahl behandelter Fälle. Erkrankungen, für die weltweit weniger als 100 Fallberichte existieren, stellen auch für große Zentren Herausforderungen dar.
Insofern scheint es sinnvoll, Kompetenzzentren für bestimmte seltene Erkrankungen zu bilden. Zudem ist ein reger Austausch zwischen den Zentren unter Berücksichtigung der gewonnenen Daten unerlässlich, um die diagnostischen und therapeutischen Möglichkeiten bei den seltenen Erkrankungen weiter zu entwickeln. Das Krankenhausstrukturgesetz aus dem Jahr 2015, welches u. a. finanzielle Zuschläge für Zentren für seltene Erkrankungen vorsieht, definiert auch deren Aufgaben, darunter die Erstellung von Registern oder Versorgungsatlanten zur Information von betroffenen Patienten. Vor allem für seltene Erkrankungen bilden Registerstudien ein wichtiges Fundament zur Datensammlung und zukünftigen statistischen Analyse von therapeutischem Nutzen, Lebensqualität, Therapiekosten sowie Arzneimittelsicherheit. Hiervon werden in absehbarer Zukunft auch Patienten mit seltenen Erkrankungen der Nase, der Nasennebenhöhlen und der vorderen Schädelbasis profitieren.
|
Entität |
Anzahl von Fällen |
Prozentzahl |
|---|---|---|
|
Pleomorphes Adenom |
73 |
23% |
|
Onkozytärer Tumor |
7 |
2% |
|
Low-grade Adenokarzinom (inkl. Azinuszellkarzinom) |
67 |
21% |
|
Mukoepidermoidkarzinom |
17 |
5% |
|
Adenoidzystisches Karzinom |
54 |
17% |
|
High-grade Adenokarzinom |
93 |
30% |
|
Summe |
311 |
100% |
|
Stadium nach Kadish |
|
|---|---|
|
A |
Tumor auf die Nasenhaupthöhle begrenzt |
|
B |
Tumorausdehnung in der Nasenhaupthöhle und den Nasennebenhöhlen |
|
C |
Tumorausdehnung über die Nasenhaupthöhle und Nasennebenhöhlen hinaus |
|
D (Erweiterung nach Morita [418]) |
Vorhandensein regionaler Metastasen oder von Fernmetastasen |
Interessenkonflikt
Die Autorinnen/Autoren geben an, dass kein Interessenkonflikt besteht.
Danksagung
Prof. Dr. Thomas Hoffmann und Prof. Dr. Jörg Lindemann für die kritische Durchsicht des Manuskripts sowie Herrn Prof. Dr. Wolfgang Pirsig für das zur Verfügung gestellte Bildmaterial und die Anregungen zum Kapitel „Fehlbildungen der Nase“. Weiter gilt mein Dank Herrn Prof. Dr. Meinrad Beer, Radiologie Universitätsklinik Ulm, für die zur Verfügung gestellten CT und MRT-Aufnahmen. Für die Bereitstellung der Abbildungen 13 und 14 bedanke ich mich bei Frau Alexandra Gey, Herrn Prof. Stefan Plontke und Dr. Alexander Glien (Klinik für Hals-Nasen-Ohrenheilkunde, Universitätsklinikum Halle) sowie bei Frau Prof. Sabrina Kösling (Radiologie, Universitätsklinikum Halle).
-
Literaturverzeichnis
- 1 Bundesministerium für Gesundheit Seltene Erkrankungen - Bundesgesundheitsministerium. 2019 Im Internet: https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
- 2 Anzahl I. Prävalenzen en und Inzidenzen seltener Krankheiten: Bibliografische Angaben. 2015; 1-52 . Im Internet: https://www.orpha.net/orphacom/cahiers/docs/DE/Pravalenzen_seltener_Krankheiten_Alphabetische_Liste.pdf
- 3 Pedan H, Janosova V, Hajtman A, Calkovsky V. Non-Reflex Defense Mechanisms of Upper Airway Mucosa: Possible Clinical Application. Physiol Res 2020; 69 (Suppl. 01) S55-S67 PMID: 32228012
- 4 Sommer F, Scheithauer M, Kröger R. et al. Niesen als mechanische Abwehr – Numerische Simulation mit Analyse der durchströmten Nasenbereiche. Laryngorhinootologie 2014; 93: 746-750
- 5 Sommer F, Hoffmann TK, Mlynski G. et al. Dreidimensionale Analyse nasaler PhysiologieThree-dimensional analysis of nasal physiology. HNO 2017; 66: 280-289
- 6 Lindemann J, Reichert M, Kröger R. et al. Numerical simulation of humidification and heating during inspiration in nose models with three different located septal perforations. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1795-1800
- 7 Sommer F, Kröger R, Lindemann J. Numerical simulation of humidification and heating during inspiration within an adult nose. Rhinology 2012; 50: 157-164
- 8 Lindemann J, Rettinger G, Kröger R. et al. Numerical simulation of airflow patterns in nose models with differently localized septal perforations. Laryngoscope 2013; 123: 2085-2089
- 9 Götte K, Hörmann K. Sinonasal malignancy: What’s new?. Orl 2004; 66: 85-97
- 10 Barnes L, Eveson JW, Reichart P. et al. World health organization classification of tumours: Pathology and genetics of head and neck tumours. Lyon: International Agency for Research on Cancer; 2005
- 11 Slinger CA, McGarry GW. Nose and sinus tumours: Red flags and referral. Br J Gen Pract 2018; 68: 247-248
- 12 Le QT, Fu KK, Kaplan M. et al. Treatment of maxillary sinus carcinoma: A comparison of the 1997 and 1977 American Joint Committee on Cancer staging systems. Cancer 1999; 86: 1700-1711
- 13 Tiwari R, Hardillo JA, Mehta D. et al. Squamous cell carcinoma of maxillary sinus. Head Neck 2000; 22: 164-169
- 14 Tiwari R, Hardillo JA, Tobi H. et al. Carcinoma of the ethmoid: Results of treatment with conventional surgery and post-operative radiotherapy. Eur J Surg Oncol 1999; 25: 401-405
- 15 Laudien M. Ausgewählte seltene rhinologische Krankheitsbilder Pathogenese – Klinik – Therapie. Laryngorhinootologie 2015; 94: S272-S287
- 16 Hauser LJ, Jensen EL, Mirsky DM. et al. Pediatric anosmia: A case series. Int J Pediatr Otorhinolaryngol 2018; 110: 135-139
- 17 Hummel T, Whitcroft KL, Andrews P. et al. Position paper on olfactory dysfunction. Rhinol J 2017; 0: 1-30
- 18 Orphanet: Anosmie, isolierte kongenitale. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=11807&Disease_Disease_Search_diseaseGroup=anosmie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Anosmie--isolierte-kongenitale&title=Anosmie
isolierte kongenit
- 19 Mahmood AN, Abulaban O, Janjua A. Bilateral olfactory aplasia: Uncommon cause of congenital anosmia. Clin Case Reports 2019; 7: 1456-1457
- 20 Orphanet: Kallmann Syndrom. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=478
- 21 Johnson VP, McMillin JM, Aceto T. et al. A newly recognized neuroectodermal syndrome of familial alopecia, anosmia, deafness, and hypogonadism. Am J Med Genet 1983; 15: 497-506
- 22 Gebril O, Abdelraouf E, Shafie M. et al. Johnson-McMillin microtia syndrome: New additional family. J Fam Med Prim Care 2014; 3: 275
- 23 Orphanet: Arrhinie, isolierte. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=DE&Expert=1134
- 24 Thiele H, Musil A, Nagel F. et al. Familial arhinia, choanal atresia, and microphthalmia. Am J Med Genet 1996; 63: 310-313
- 25 Abukhalaf SA, Zalloum JS, Al Hammouri A. et al. Congenital arrhinia: A case report and literature review. Int J Pediatr Otorhinolaryngol 2020; 135: 110083
- 26 Jung JW, Ha DH, Kim BY. et al. Nasal Reconstruction Using a Customized Three-Dimensional–Printed Stent for Congenital Arhinia: Three-Year Follow-up. Laryngoscope 2019; 129: 582-585
- 27 Núñez-Villaveiran T, Frohner BB, Urcelay PR. et al. Bifid nose – A mild degree of frontonasal dysplasia. A case report. Int J Pediatr Otorhinolaryngol 2013; 77: 1374-1377
- 28 Orphanet: Bifid nose. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2457&Disease_Disease_Search_diseaseGroup=bifid-nose&Disease_Disease_Search_diseaseType=Pat&Disease(s)/groupofdiseases=Bifid-nose&title=Bifid
nose&search=Disease_Search_Simple
- 29 Sedano HO, Michael Cohen M, Jirasek J. et al. Frontonasal dysplasia. J Pediatr 1970; 76: 906-913
- 30 Genç E, Derbent M, Ergin NT. A mild case of frontonasal dysplasia: The rhinologic perspective. Int J Pediatr Otorhinolaryngol 2002; 65: 75-83
- 31 Cohen MM. Perspectives on craniosynostosis: Sutural biology, some well-known syndromes, and some unusual syndromes. J Craniofac Surg 2009; 20: 646-651
- 32 Orphanet: Kraniorhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=de&Expert=157832
- 33 Rüegg EM, Bartoli A, Rilliet B. et al. Management of median and paramedian craniofacial clefts. J Plast Reconstr Aesthetic Surg 2019; 72: 676-684
- 34 Orphanet: Paramedian nasal cleft. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=17067&Disease_Disease_Search_diseaseGroup=paramediannasal-cleft&Disease_Disease_Search_diseasType=Pat&Disease(s)/groupofdiseases=Paramedian-nasal-cleft&title=Paramedian%20nasal%20cleft
- 35 Williams A, Pizzuto M, Brodsky L. et al. Supernumerary nostril: A rare congenital deformity. Int J Pediatr Otorhinolaryngol 1998; 44: 161-167
- 36 Orphanet: Polyrhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17033&Disease_Disease_Search_diseaseGroup=Polyrhinie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Polyrhinie&title=Polyrhinie&search=Disease_Search_Simple
- 37 Orphanet: Nasenlöcher, überzählige. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17034&Disease_Disease_Search_diseaseGroup=Nasenloch-&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Nasenlocher--uberzahlige&title=Nasenl%F6cher,%FCberz%E4hlige
- 38 Gulasi S, Turhan AH, Celik Y. et al. Accessory nostril: A rare congenital nasal anomaly. J Craniofac Surg 2015; 26: e602-e603
- 39 Galiè M, Clauser LC, Tieghi R. et al. The arrhinias: Proboscis lateralis literature review and surgical update. J Cranio-Maxillofacial Surg 2019; 47: 1410-1413
- 40 Harada T, Muraoka M. Proboscis lateralis: A rare bilateral case [7]. Ann Plast Surg 2001; 47: 350-351
- 41 Wolfe SA, Tessier P, Ciminello FS. The Arrhinias. Scand J Plast Reconstr Surg Hand Surg 2009; 43: 177-196
- 42 Rosen Z, Gitlin G. Bilateral Nasal Proboscis: Associated with Unilateral Anophthalmia, Unilateral Diffuse Pigmentation of the Conjunctiva, and Anomalies of the Skull and Brain. AMA Arch Otolaryngol 1959; 70: 545-550
- 43 El-fattah AMA, Naguib A, El-Sisi H. et al. Midline nasofrontal dermoids in children: A review of 29 cases managed at Mansoura University Hospitals. Int J Pediatr Otorhinolaryngol 2016; 83: 88-92
- 44 Orphanet: Nasal dorsum fistula. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=141219
- 45 Jurkov M, Olze H, Klauschen F. et al. IgG4-related orbitopathy as an important differential diagnosis of advanced silent sinus syndrome. HNO 2020; 68: 65-68
- 46 MONTGOMERY WW. Mucocele of the Maxillary Sinus Causing Enophthalmos. Eye Ear Nose Throat Mon 1964; 43: 41-44
- 47 Soparkar CNS, Patrinely JR, Cuaycong MJ. et al. The Silent Sinus Syndrome: A Cause of Spontaneous Enophthalmos. Ophthalmology 1994; 101: 772-778
- 48 Naik RM, Khemani S, Saleh HA. Frontal silent sinus syndrome. Otolaryngol - Head Neck Surg (United States) 2013; 148: 354-355
- 49 McArdle B, Perry C. Ethmoid silent sinus syndrome causing inward displacement of the orbit: Case report. J Laryngol Otol 2010; 124: 206-208
- 50 Wise SK, Wojno TH, DelGaudio JM. Silent sinus syndrome: Lack of orbital findings in early presentation. Am J Rhinol 2007; 21: 489-494
- 51 Brandt MG, Wright ED. The silent sinus syndrome is a form of chronic maxillary atelectasis: A systematic review of all reported cases. Am J Rhinol 2008; 22: 68-73
- 52 Rose GE, Lund VJ. Clinical features and treatment of late enophthalmos after orbital decompression: A condition suggesting cause for idiopathic „imploding antrum“ (silent sinus) syndrome. Ophthalmology 2003; 110: 819-826
- 53 Numa WA, Desai U, Gold DR. et al. Silent sinus syndrome: A case presentation and comprehensive review of all 84 reported cases. Ann Otol Rhinol Laryngol 2005; 114: 688-694
- 54 Davidson JK, Soparkar CNS, Williams JB. et al. Negative sinus pressure and normal predisease imaging in silent sinus syndrome. Arch Ophthalmol 1999; 117: 1653-1654
- 55 Annino DJ, Goguen LA. Silent sinus syndrome. Curr Opin Otolaryngol Head Neck Surg 2008; 16: 22-25
- 56 Hildenbrand T, Klein SB, Schramek N. et al. Rare diseases of the maxillary sinus. HNO 2020; 68: 581-589
- 57 Facon F, Eloy P, Brasseur P. et al. The silent sinus syndrome. Eur Arch Oto-Rhino-Laryngology 2006; 263: 567-571
- 58 Hourany R, Aygun N, Della Santina CC. et al. Silent sinus syndrome: An acquired condition. Am J Neuroradiol 2005; 26: 2390-2392
- 59 Lee DS, Murr AH, Kersten RC. et al. Silent sinus syndrome without opacification of ipsilateral maxillary sinus. Laryngoscope 2018; 128: 2004-2007
- 60 Illner A, Davidson HC, Harnsberger HR. et al. The silent sinus syndrome: Clinical and radiografic findings. Am J Roentgenol 2002; 178: 503-506
- 61 Rose GE, Sandy C, Hallberg L. et al. Clinical and radiologic characteristics of the imploding antrum, or „silent sinus,“ syndrome. Ophthalmology 2003; 110: 811-818
- 62 Weber RK. Aktueller Stand der endonasalen Nasennebenhöhlenchirurgie
[Comprehensive review on endonasal endoscopic sinus
surgery]. Laryngorhinootologie. 2015; 94 Suppl 1; S64–S142.
doi:110.1055/s-0035-1545353. Epub 2015 Apr 10. PMID: 25860497
- 63 Sogg A. Endoscopic sinus surgery. 4th Editio Thieme Verlag; 1993
- 64 Sommer F, Hoffmann T, Lindemann J. et al. Radicality of maxillary sinus surgery and size of the maxillary sinus ostium. HNO 2020; 68: 573-580
- 65 Thomas RD, Graham SM, Carter KD. et al. Management of the orbital floor in silent sinus syndrome. Am J Rhinol 2003; 17: 97-100
- 66 Babar-Craig H, Kayhanian H, de Silva DJ. et al. Spontaneous silent sinus syndrome (Imploding antrum syndrome): Case series of 16 patients. Rhinology 2011; 49: 315-317
- 67 Sivasubramaniam R, Sacks R, Thornton M. Silent sinus syndrome: Dynamic changes in the position of the orbital floor after restoration of normal sinus pressure. J Laryngol Otol 2011; 125: 1239-1243
- 68 Adams WM, Jones RI, Chavda SI. et al. Pneumosinus dilatans: a discussion of four cases and the possible aetiology. Rhinology 1998; 36: 40-402
- 69 Urken ML, Som PM, Lawson W. et al. Abnormally large frontal sinus. II. Nomenclature, pathology, and symptoms. Laryngoscope 1987; 97: 606-611
- 70 Kim WJ, Kim MM. Binasal hemianopia caused by pneumosinus dilatans of the sphenoid sinuses. Indian J Ophthalmol 2019; 67: 1772-1775
- 71 Skolnick CA, Mafee MF, Goodwin JA. Pneumosinus Dilatans of the Sphenoid Sinus Presenting With Visual Loss. J Neuro-ophthalmology 2001; 20: 259-263
- 72 Alatar AA, AlSuliman YA, Alrajhi MS. et al. Maxillary Pneumosinus Dilatans Presenting With Proptosis: A Case Report and Review of the Literature. Clin Med Insights Ear, Nose Throat 2019; 12: 117955061882514
- 73 Sweatman J, Beltechi R. Pneumosinus Dilatans: An exploration into the association between Arachnoid Cyst, Meningioma and the pathogenesis of Pneumosinus Dilatans. Clin Neurol Neurosurg 2019; 185
- 74 Ricci JA. Pneumosinus Dilatans: Over 100 Years Without an Etiology. J Oral Maxillofac Surg 2017; 75: 1519-1526
- 75 Martin AJ, Jarosz JM, Thomas NWM. The strange association of pneumosinus dilatans and arachnoid cyst: Case report and review of the literature. Acta Neurochir (Wien) 2001; 143: 197-201
- 76 Gibbons BA, Miele WR, Florman JE, Heilman CB, Horgan MA
Pneumosinus dilitans and meningioma: a case series and review of
the literature. Neurosurg Focus 2011; 50(5): E13. doi:10.3171/2011.
3.FOCUS1113. PMID: 21529169
- 77 Desai NS, Saboo SS, Khandelwal A. et al. Pneumosinus dilatans: Is it more than an aesthetic concern?. J Craniofac Surg 2014; 25: 418-421
- 78 Draf W, Constantinidis J, Weber R. et al. Pneumosinus Dilatans Frontalis, Atiologie, Symptomatik Und Operationstechnik. Laryngorhinootologie 1996; 75: 660-664
- 79 Cho HS, Hong SJ, Chae HK. et al. Maxillary Sinus Pneumocele Presenting as Aesthetic Deformity: A Case Report With Literature Review. Ear, Nose Throat J 2020; 99: 397-401
- 80 Song M, Ahn SM, Reh DR. et al. Ethmoid pneumocele presenting with exophthalmos 15 years after endoscopic sinus surgery. Allergy Rhinol 2015; 6: 129-132
- 81 Braverman I. Pneumocele of the maxillary sinus with orbital and
trigeminal nerve involvement: case report and review of the
literature. J Otolaryngol Head Neck Surg 2009; 38(2): E35–E38.
PMID: 19442351
- 82 Sasaki T, Yamoto T, Fujita K. et al. A Case of Orbital Emphysema Associated with Frontal Sinus Pneumocele. J Neurol Surg Reports 2013; 74: 054-056
- 83 Bachor E, Weber R, Kahle G. et al. Temporary unilateral amaurosis with pneumosinus dilatans of the sphenoid sinus. Skull Base Surg 1994; 4: 169-175
- 84 Bell AF, Ivan DJ, Munson RA. Barosinus pneumocele: Transient visual loss due to sphenoid sinus pneumocele in a U.S. Air Force pilot. Aviat Sp Environ Med 1995; 66: 276-279
- 85 Choi SJ, Seo ST, Rha KS. et al. Sinonasal organized Hematoma: Clinical features of seventeen cases and a systematic review. Laryngoscope 2015; 125: 2027-2033
- 86 Kim JS, Oh JS, Kwon SH. The increasing incidence of paranasal organizing hematoma: a 20-year experience of 23 cases at a single center. Rhinol J 2016; 54: 176-182
- 87 Pang W, Hu L, Wang H. et al. Organized Hematoma: An Analysis of 84 Cases with Emphasis on Difficult Prediction and Favorable Management. Otolaryngol – Head Neck Surg (United States) 2016; 154: 626-633
- 88 Song HM, Jang YJ, Chung YS. et al. Organizing hematoma of the maxillary sinus. Otolaryngol - Head Neck Surg 2007; 136: 616-620
- 89 Young D. Surgical treatment of male infertility. J Reprod Fertil 1970; 23: 541-542
- 90 Hendry WF, Knight RK, Whitfield HN. et al. Obstructive Azoospermia: Respiratory Function Tests, Electron Microscopy and the Results of Surgery. Br J Urol 1978; 50: 598-604
- 91 Arya AK, Beer HL, Benton J. et al. Does Young’s syndrome exist?. J Laryngol Otol 2009; 123: 477-481
- 92 Hendry WF, Levison DA, Parkinson MC. et al. Testicular obstruction: Clinicopathological studies. Ann R Coll Surg Engl 1990; 72: 396-407
- 93 Shiraishi K, Ono N, Eguchi S. et al. Young’s syndrome associated with situs inversus totalis. Arch Androl 2004; 50: 169-172
- 94 Hasegawa A, Fujita M, Ohe M. et al. A Rare Case of Young’s Syndrome in Japan. Intern Med 1994; 33: 649-653
- 95 Armengot M, Juan G, Carda C. et al. Young’s syndrome: A further cause of chronic rhinosinusitis. Rhinology 1996; 34: 35-37
- 96 Greenstone MA, Rutman A, Hendry WF. et al. Ciliary function in Young’s syndrome. Thorax 1988; 43: 153-154
- 97 Stern BM, Sharma G. Ciliary Dysfunction (Kartagener Syndrome, Primary Ciliary Dyskinesia). StatPearls Publishing; 2019
- 98 Samia H, Khadija B, Agnes H. et al. Long-term outcome of tunisian children with primary ciliary dyskinesia confirmed by transmission electron microscopy. Afr Health Sci 2016; 16: 954-961
- 99 Schofield LM, Duff A, Brennan C. Airway Clearance Techniques for Primary Ciliary Dyskinesia; is the Cystic Fibrosis literature portable?. Paediatr Respir Rev 2018; 25: 73-77
- 100 Alanin MC, Aanaes K, Høiby N. et al. Sinus surgery can improve quality of life, lung infections, and lung function in patients with primary ciliary dyskinesia. Int Forum Allergy Rhinol 2017; 7: 240-247
- 101 Nyilas S, Schlegtendal A, Singer F, Goutaki M, Kuehni CE, Casaulta C,
Latzin P, Koerner-Rettberg C. Alternative inert gas washout outcomes
in patients with primary ciliary dyskinesia. Eur Respir J 2017; 49(1):
1600466. doi:10.1183/13993003.00466-2016. PMID: 28122863
- 102 Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, Dell S,
Eber E, Escudier E, Hirst RA, Hogg C, Jorissen M, Latzin P, Legendre M,
Leigh MW, Midulla F, Nielsen KG, Omran H, Papon JE, Pohunek P,
Redfern B, Rigau D, Rindlisbacher B, Santamaria F, Shoemark A,
Snijders D, Tonia T, Titieni A, Walker WT, Werner C, Bush A, Kuehni
CE. European Respiratory Society guidelines for the diagnosis of
primary ciliary dyskinesia. Eur Respir J 2017; 49(1); 1601090.
doi:10.1183/13993003.00466-2016. PMID: PMC6054534
- 103 Strippoli MPF, Frischer T, Barbato A. et al. Management of primary ciliary dyskinesia in European children: Recommendations and clinical practice. Eur Respir J 2012; 39: 1482-1491
- 104 Barbato A, Frischer T, Kuehni CE. et al. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. In: European Respiratory Journal. Eur Respir J 2009; 1264-1276
- 105 Lucas JS, Burgess A, Mitchison HM. et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child 2014; 99: 850-856
- 106 El-Naggar AK, Chan JKC, Grandis JR et al. WHO Classification of Head
and Neck Tumours. 4th Edition Lyon: International Agency for
Research on Cancer; 2017
- 107 Lombardi D, Tomenzoli D, Buttà L. et al. Limitations and complications of endoscopic surgery for treatment for sinonasal inverted papilloma: A reassessment after 212 cases. Head Neck 2011; 33: 1154-1161
- 108 Stange T, Schultz-Coulon HJ. Zum Chirurgischen Behandlungskonzept bei Invertierten Papillomen der Nase und Nasennebenhöhlen. HNO 2008; 56: 614-622
- 109 Christensen WN, Smith RRL. Schneiderian papillomas. A clinicopathologic study of 67 cases. Hum Pathol 1986; 17: 393-400
- 110 Stammberger H. New aspects in the genesis of inverted papillomas. Laryngol Rhinol Otol (Stuttg) 1983; 62: 249-24955
- 111 Leoncini G, Zanetti L. The papillomas of the sinonasal tract. A comprehensive review. Pathologica 2017; 109: 31-34
- 112 Sarkar FH, Visscher DW, Kintanar EB. et al. Sinonasal Schneiderian papillomas: human papillomavirus typing by polymerase chain reaction. Mod Pathol 1992; 5: 329-332
- 113 Barnes L. Schneiderian papillomas and nonsalivary glandular neoplasms of the head and neck. Mod Pathol 2002; 15: 279-297
- 114 Cunningham MJ, Brantley S, Barnes L. et al. Oncocytic Schneiderian papilloma in a young adult: A rare diagnosis. Otolaryngol Neck Surg 1987; 97: 47-51
- 115 Barnes L, Bedetti C. Oncocytic schneiderian papilloma: A reappraisal of cylindrical cell papilloma of the sinonasal tract. Hum Pathol 1984; 15: 344-351
- 116 Michaels L, Young M. Histogenesis of papillomas of the nose and paranasal sinuses. Arch Pathol Lab Med 1995; 119: 821-826
- 117 Lawson W, Patel ZM. The evolution of management for inverted papilloma: An analysis of 200 cases. Otolaryngol – Head Neck Surg 2009; 140: 330-335
- 118 Lund VJ, Stammberger H, Nicolai P. et al. European position paper on endoscopic management of tumours of the nose, paranasal sinuses and skull base. Rhinol Suppl 2010; 1-143
- 119 Maitra A, Baskin LB, Lee EL. Malignancies arising in oncocytic schneiderian papillomas: A report of 2 cases and review of the literature. Arch Pathol Lab Med 2001; 125: 1365-1367
- 120 Anari S, Carrie S. Sinonasal inverted papilloma: Narrative review. J Laryngol Otol 2010; 124: 705-715
- 121 Attlmayr B, Derbyshire SG, Kasbekar AV et al. Management of
inverted papilloma: Review. In: Journal of Laryngology and Otology.
Cambridge University Press; 2017: 284–289
- 122 Heathcote KJ, Nair SB. The impact of modern techniques on the recurrence rate of inverted papilloma treated by endonasal surgery. Rhinology 2009; 47: 339-344
- 123 Diamantopoulos II, Jones NS, Lowe J. All nasal polyps need histological examination: An audit-based appraisal of clinical practice. J Laryngol Otol 2000; 114: 755-759
- 124 Lloyd GAS, Lloyd GAS. Epithelial Tumours. In: Diagnostic Imaging of the Nose and Paranasal Sinuses 1988; 95-110
- 125 Hildenbrand T, Weber R, Mertens J. et al. Surgery of Inverted Papilloma of the Maxillary Sinus via Translacrimal Approach – Long-Term Outcome and Literature Review. J Clin Med 2019; 8: 1873
- 126 Giotakis E, Knipping S, Kühnel T. et al. Unilateral diseases of the maxillary sinus. HNO 2020; 68: 623-636
- 127 Vrabec DP. The inverted schneiderian papilloma: A 25-year study. Laryngoscope 1994; 104: 582-605
- 128 Bielamowicz S, Calcaterra TC, Watson D. Inverting papilloma of the head and neck: The UCLA update. Otolaryngol – Head Neck Surg 1993; 109: 71-76
- 129 Kraft M, Simmen D, Kaufmann T. et al. Long-term results of endonasal sinus surgery in sinonasal papillomas. Laryngoscope 2003; 113: 1541-1547
- 130 Tanna N, Edwards JD, Aghdam H. et al. Transnasal endoscopic medial maxillectomy as the initial oncologic approach to sinonasal neoplasms: The anatomic basis. Arch Otolaryngol – Head Neck Surg 2007; 133: 1139-1142
- 131 Busquets JM, Hwang PH. Endoscopic resection of sinonasal inverted papilloma: A meta-analysis. Otolaryngol – Head Neck Surg 2006; 134: 476-482
- 132 Veeresh M, Sudhakara M, Girish G. et al. Leiomyoma: A rare tumor in the head and neck and oral cavity: Report of 3 cases with review. J Oral Maxillofac Pathol 2013; 17: 281-287
- 133 Agaimy A, Michal M, Thompson LDR. et al. Angioleiomyoma of the Sinonasal Tract: Analysis of 16 Cases and Review of the Literature. Head Neck Pathol 2015; 9: 463-473
- 134 Heyman JNS, Jones LM, Hilton JM. et al. Angioleiomyoma of the inferior turbinate: A rare cause of isolated facial pain. Ann R Coll Surg Engl 2020; 102: E20-E22
- 135 Lau YW, Tharumalingam V, Tan TY. et al. Sinonasal angioleiomyoma. Med J Malaysia 2016; 71: 154-155
- 136 Mathieu T, Verbruggen A, Goovaerts G. et al. Vascular leiomyoma of the nasal cavity: case report and literature review. Eur Arch Oto-Rhino-Laryngology 2017; 274: 579-583
- 137 Poncet A, Dor L. Botryomycose humaine. Rev Chir 1897; 18: 996-1003
- 138 Hartzell M. Granuloma pyogenicum (botryomycosis of French authors). J Cutan Dis 1904; 22: 520-523
- 139 Mills SE, Cooper PH, Fechner RE. Lobular capillary hemangioma: The underlying lesion of pyogenic granuloma. A study of 73 cases from the oral and nasal mucous membranes. Am J Surg Pathol 1980; 4: 471-479
- 140 Fu Y-S, Perzin KH. Nonepithelial tumors of the nasal cavity, paranasal sinuses, and nasopharynx. A clinicopathologic study VI. Fibrous tissue tumors (fibroma, fibromatosis, fibrosarcoma). Cancer 1976; 37: 2912-2928
- 141 Thompson LDR, Fanburg-Smith JC. Update on Select Benign Mesenchymal and Meningothelial Sinonasal Tract Lesions. Head Neck Pathol 2016; 10: 95-108
- 142 Sammut SJ, Tomson N, Corrie P. Pyogenic granuloma as a cutaneous adverse effect of vemurafenib. N Engl J Med 2014; 371: 1265-1267
- 143 Smith SC, Patel RM, Lucas DR. et al. Sinonasal Lobular Capillary Hemangioma: A Clinicopathologic Study of 34 Cases Characterizing Potential for Local Recurrence. Head Neck Pathol 2013; 7: 129-134
- 144 Sunaryo PL, Svider PF, Husain Q. et al. Schwannomas of the sinonasal tract and anterior skull base: A systematic review of 94 cases. Am J Rhinol Allergy 2014; 28: 39-49
- 145 Shugar JMA, Son PM, Biller HF. et al. Peripheral nerve sheath tumors of the paranasal sinuses. Head Neck Surg 1981; 4: 72-76
- 146 Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg 1994; 81: 362-373
- 147 Ansari I, Ansari A, Graison AA. et al. Head and Neck Schwannomas: A Surgical Challenge – A Series of 5 Cases. Case Rep Otolaryngol 2018; 2018: 1-10
- 148 Azani AB, Bishop JA, Thompson LDR. Sinonasal Tract Neurofibroma: A Clinicopathologic Series of 12 Cases with a Review of the Literature. Head Neck Pathol 2015; 9: 323-333
- 149 Annino DJ, Domanowski GF, Vaughan CW. A Rare Cause of Nasal Obstruction: A Solitary Neurofibroma. Otolaryngol Neck Surg 1991; 104: 484-488
- 150 Perzin KH, Panyu H, Wechter S. Nonepithelial tumors of the nasal cavity, paranasal sinuses and nasopharynx. A clinicopathologic study. XII: Schwann cell tumors (neurilemoma, neurofibroma, malignant schwannoma). Cancer 1982; 50: 2193-2202
- 151 Friedman CD, Costantino PD, Teitelbaum B. et al. Primary extracranial meningiomas of the head and neck. Laryngoscope 1990; 100: 41-48
- 152 Thompson LDR, Gyure KA. Extracranial sinonasal tract meningiomas: A clinicopathologic study of 30 cases with a review of the literature. Am J Surg Pathol 2000; 24: 640-650
- 153 Ho K-L. Primary meningioma of the nasal cavity and paranasal sinuses. Cancer 1980; 46: 1442-1447
- 154 Perry A, Scheithauer BW, Stafford SL. et al. „Malignancy“ in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer 1999; 85: 2046-2056
- 155 Perry A, Stafford SL, Scheithauer BW. et al. Meningioma grading: An analysis of histologic parameters. Am J Surg Pathol 1997; 21: 1455-1465
- 156 Rushing EJ, Bouffard JP, McCall S. et al. Primary extracranial meningiomas: An analysis of 146 cases. Head Neck Pathol 2009; 3: 116-130
- 157 Mnejja M, Hammami B, Bougacha L. et al. Primary sinonasal meningioma. Eur Ann Otorhinolaryngol Head Neck Dis 2012; 129: 47-50
- 158 Wolf A, Safran B, Pock J. et al. Surgical treatment of paranasal sinus osteomas: A single center experience of 58 cases. Laryngoscope. 2019
- 159 Atallah N, Jay MM. Osteomas of the paranasal sinuses. J Laryngol Otol 1981; 95: 291-304
- 160 Furlaneto EC, Rocha JRM, Heitz C. Osteoma of the zygomatic arch - Report of a case. Int J Oral Maxillofac Surg 2004; 33: 310-311
- 161 Summers LE, Mascott CR, Tompkins JR. et al. Frontal sinus osteoma associated with cerebral abscess formation: A case report. Surg Neurol 2001; 55: 235-239
- 162 Eller R, Sillers M. Common Fibro-osseous Lesions of the Paranasal Sinuses. Otolaryngol Clin North Am 2006; 39: 585-600
- 163 Schick B, Steigerwald C, El Tahan AER. et al. The role of endonasal surgery in the management of frontoethmoidal osteomas. Rhinology 2001; 39: 66-70
- 164 Schick B, Dlugaiczyk J. Benign tumors of the nasal cavity and paranasal sinuses. In: Rhinology and Facial Plastic Surgery 2009; 377-385
- 165 Buyuklu F, Akdogan MV, Ozer C. et al. Growth characteristics and clinical manifestations of the paranasal sinus osteomas. Otolaryngol - Head Neck Surg 2011; 145: 319-323
- 166 Golant A, Zeichner JA. Gardner Syndrome. StatPearls Publishing; 2014
- 167 Pinto RS, Simons A, Verma R. et al. Gardener-associated fibroma: An unusual cause of upper airway obstruction. BMJ Case Rep 2018; 2018
- 168 Yang A, Sisson R, Gupta A, Tiao G, Geller JI. Germline APC mutations
in hepatoblastoma. Pediatr Blood Cancer 2018; 65(4); doi:10.1002/
pbc.26892. Epub 2017 Dec 18. PMID: 29251405
- 169 Kiessling P, Dowling E, Huang Y, Ho ML, Balakrishnan K, Weigel BJ,
Highsmith WE Jr, Niu Z, Schimmenti LA. Identification of aggressive
Gardner syndrome phenotype associated with a de novo APC variant,
c.4666dup. Cold Spring Harb Mol Case Stud 2019; 5(2): a003640.
doi:10.1101/mcs.a003640. PMID: 30696621; PMCID: PMC6549566
- 170 Burke AB, Collins MT, Boyce AM. Fibrous dysplasia of bone: craniofacial and dental implications. Oral Dis 2017; 23: 697-708
- 171 Erkmen K, Al-Mefty O, Adada B. Tumors of the skull base. In: Oncology of CNS Tumors Philadelphia: WB Saunders; 2010: 279-307
- 172 Heller AJ, DiNardo LJ, Massey D. Fibrous dysplasia, chondrosarcoma, and McCune-Albright syndrome. Am J Otolaryngol – Head Neck Med Surg 2001; 22: 297-301
- 173 Kos M, Luczak K, Godzinski J. et al. Treatment of monostotic fibrous dysplasia with pamidronate. J Cranio-Maxillofacial Surg 2004; 32: 10-15
- 174 Collins MT, Kushner H, Reynolds JC. et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res 2005; 20: 219-226
- 175 Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: Age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int 2008; 19: 57-63
- 176 Devogelaer JP. New uses of bisphosphonates: Osteogenesis imperfecta. Curr Opin Pharmacol 2002; 2: 748-753
- 177 Schreiber A, Villaret AB, Maroldi R. et al. Fibrous dysplasia of the sinonasal tract and adjacent skull base. Curr Opin Otolaryngol Head Neck Surg 2012; 20: 45-52
- 178 Amit M, Collins MT, FitzGibbon EJ. et al. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia – a meta-analysis. PLoS One 2011; 6
- 179 McCune D. Osteitis fibrosa cystica: The case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 1936; 52: 743-744
- 180 Leet AI, Chebli C, Kushner H. et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res 2004; 19: 571-577
- 181 Boyce AM, Brewer C, Deklotz TR. et al. Association of hearing loss and otologic outcomes with fibrous dysplasia. JAMA Otolaryngol – Head Neck Surg 2018; 144: 102-107
- 182 Deklotz TR, Kim HJ, Kelly M. et al. Sinonasal disease in polyostotic fibrous dysplasia and McCune-Albright Syndrome. Laryngoscope 2013; 123: 823-828
- 183 Boyce AM, Glover M, Kelly MH. et al. Optic neuropathy in McCune-Albright syndrome: Effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab 2013; 98
- 184 Plotkin H, Rauch F, Zeitlin L. et al. Effect of Pamidronate Treatment in Children with Polyostotic Fibrous Dysplasia of Bone. J Clin Endocrinol Metab 2003; 88: 4569-4575
- 185 DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev 2007; 4: 440-445
- 186 Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; (03) 12 PMID: 18489744 PMCID: PMC2459161
- 187 Feldman MD, Kelly M, Rao VM. et al. Fibrous dysplasia of the paranasal sinuses. Otolaryngol Neck Surg 1986; 95: 222-225
- 188 Ikeda K, Suzuki H, Oshima T. et al. Endonasal endoscopic management in fibrous dysplasia of the paranasal sinuses. Am J Otolaryngol – Head Neck Med Surg 1997; 18: 415-418
- 189 Uzun C, Adali MK, Koten M. et al. McCune-Albright syndrome with fibrous dysplasia of the paranasal sinuses. Rhinology 1999; 37: 122-124
- 190 Lee JS, Fitzgibbon E, Butman JA. et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 2002; 347: 1670-1676
- 191 Wenig BM, Heffner DK. Respiratory epithelial adenomatoid hamartomas of the sinonasal tract and nasopharynx: A clinicopathologic study of 31 cases. Ann Otol Rhinol Laryngol 1995; 104: 639-645
- 192 Delbrouck C, Fernandez Aguilar S, Choufani G. et al. Respiratory epithelial adenomatoid hamartoma associated with nasal polyposis. Am J Otolaryngol – Head Neck Med Surg 2004; 25: 282-284
- 193 Endo R, Matsuda H, Takahashi M. et al. Respiratory epithelial adenomatoid hamartoma in the nasal cavity. Acta Otolaryngol 2002; 122: 398-400
- 194 Ingram WF, Noone MC, Gillespie MB. Respiratory epithelial adenomatoid hamartoma: A case report. Ear, Nose Throat J 2006; 85: 190-192
- 195 Athre R, Ducic Y. Frontal sinus hamartomas. Am J Otolaryngol – Head Neck Med Surg 2005; 26: 419-421
- 196 Flavin R, Russell J, Phelan E. et al. Chondro-osseous respiratory epithelial adenomatoid hamartoma of the nasal cavity: A case report. Int J Pediatr Otorhinolaryngol 2005; 69: 87-91
- 197 Fitzhugh VA, Mirani N. Respiratory epithelial adenomatoid hamartoma: A review. Head Neck Pathol 2008; 2: 203-208
- 198 Braun H, Beham A, Stammberger H. Respiratorisches epitheliales adenomatoides hamartom der nasenhaupthöhle: Fallbericht und literaturübersicht. Laryngorhinootologie 2003; 82: 416-420
- 199 Himi Y, Yoshizaki T, Sato K. et al. Respiratory epithelial adenomatoid hamartoma of the maxillary sinus. J Laryngol Otol 2002; 116: 317-318
- 200 Safi C, Li C, Tabaee A. et al. Outcomes and imaging findings of respiratory epithelial adenomatoid hamartoma: a systematic review. Int Forum Allergy Rhinol 2019; 9: 674-680
- 201 Lima NB, Jankowski R, Georgel T. et al. Respiratory adenomatoid hamartoma must be suspected on CT-scan enlargement of the olfactory clefts. Rhinology 2006; 44: 264-269
- 202 Hawley KA, Ahmed M, Sindwani R. CT findings of sinonasal respiratory epithelial adenomatoid hamartoma: A closer look at the olfactory clefts. Am J Neuroradiol 2013; 34: 1086-1090
- 203 Metselaar RM, Stel HV, Van Der Baan S. Respiratory epithelial adenomatoid hamartoma in the nasopharynx. J Laryngol Otol 2005; 119: 476-478
- 204 Hawley KA, Pabon S, Hoschar AP. et al. The presentation and clinical significance of sinonasal respiratory epithelial adenomatoid hamartoma (REAH). Int Forum Allergy Rhinol 2013; 3: 248-253
- 205 Schafer DR, Thompson LDR, Smith BC. et al. Primary ameloblastoma of the sinonasal tract: A clinicopathologic study of 24 cases. Cancer 1998; 82: 667-674
- 206 Shahidi S, Bronoosh P, Daneshbod Y. Follicular ameloblastoma presenting as a sinonasal tumor. Iran Red Crescent Med J 2012; 14: 113-116
- 207 Hertog D, van der Waal I. Ameloblastoma of the jaws: A critical reappraisal based on a 40-years single institution experience. Oral Oncol 2010; 46: 61-64
- 208 Barrena BG, Phillips BJ, Moran KJ. et al. Sinonasal Ameloblastoma. Head Neck Pathol 2019; 13: 247-250
- 209
Golbin DA, Ektova AP, Demin MO, Lasunin N, Cherekaev VA. Nasal
Chondromesenchymal Hamartoma with Skull Base and Orbital
Involvement: Case Presentation. Cureus 2018; 10(6): e2892.
doi:10.7759/cureus.2892. PMID: 30167348; PMCID: PMC6112910
- 210 McDermott MB, Ponder TB, Dehner LP. Nasal chondromesenchymal hamartoma: An upper respiratory tract analogue of the chest wall mesenchymal hamartoma. Am J Surg Pathol 1998; 22: 425-433
- 211 Ozolek JA, Carrau R, Barnes EL. et al. Nasal chondromesenchymal hamartoma in older children and adults: Series and immunohistochemical analysis. Arch Pathol Lab Med 2005; 129: 1444-1450
- 212 Chan TY. World Health Organization classification of tumours: Pathology & genetics of tumours of the urinary system and male genital organs. Urology 2005; 65: 214-215
- 213 Seibert RW, Seibert JJ, Jimenez JF. et al. Nasopharyngeal brain heterotopia – a cause of upper airway obstruction in infancy. Laryngoscope 1984; 94: 818-819
- 214 Kanjanawasee D, Chaowanapanja P, Keelawat S. et al. Sphenoid Sinus Cholesteatoma – Complications and Skull Base Osteomyelitis: Case Report and Review of Literature. Clin Med Insights Case Reports 2019; 12
- 215 Hansen S, Sørensen CH, Stage J. et al. Massive cholesteatoma of the frontal sinus: Case report and review of the literature. Auris Nasus Larynx 2007; 34: 387-392
- 216
Gey A, Plontke SK, Scheller C, Kösling S, Fathke C, Glien A. Seltene
Diagnose einer knochendestruierenden Läsion der Keilbeinhöhle
[Rare diagnosis of a bone-destructive lesion of the sphenoid sinus].
HNO. 2020 Dec 14. German. doi:10.1007/s00106-020-00974-2.
Epub ahead of print. PMID: 33315128
- 217 Ansa B, Goodman M, Ward K. et al. Paranasal sinus squamous cell carcinoma incidence and survival based on surveillance, epidemiology, and end results data, 1973 to 2009. Cancer 2013; 119: 2602-2610
- 218 Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: A historical analysis of population-based data. Head Neck 2012; 34: 877-885
- 219 Sanghvi S, Khan MN, Patel NR. et al. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4994 patients. Laryngoscope 2014; 124: 76-83
- 220 Youlden DR, Cramb SM, Peters S. et al. International comparisons of the incidence and mortality of sinonasal cancer. Cancer Epidemiol 2013; 37: 770-779
- 221 Hayes RB, Kardaun JWPF, De Bruyn A. Tobacco use and sinonasal cancer: A case-control study. Br J Cancer 1987; 56: 843-846
- 222 Brinton LA, Blot WJ, Becker JA. et al. A case-control study of cancers of the nasal cavity and paranasal sinuses. Am J Epidemiol 1984; 119: 896-906
- 223 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian Papilloma (Malignant Transformation): A Clinicopathologic and Immunophenotypic Study of 20 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2014; 8: 269-286
- 224 Dulguerov P, Jacobsen M, Allal A. et al. Nasal and paranasal sinus carcinoma: are we making progress? A series of 220 patients and a systematic review. Cancer 2001; 92: 3012-3029
- 225 Thorup C, Sebbesen L, Danø H. et al. Carcinoma of the nasal cavity and paranasal sinuses in Denmark 1995-2004. Acta Oncol (Madr) 2010; 49: 389-394
- 226 El-Mofty SK, Lu DW. Prevalence of high-risk human papillomavirus DNA in nonkeratinizing (cylindrical cell) carcinoma of the sinonasal tract: A distinct clinicopathologic and molecular disease entity. Am J Surg Pathol 2005; 29: 1367-1372
- 227 Bishop JA, Guo TW, Smith DF. et al. Human papillomavirus-related carcinomas of the sinonasal tract. Am J Surg Pathol 2013; 37: 185-192
- 228 Osborn DA. Nature and behavior of transitional tumors in the upper respiratory tract. Cancer 1970; 25: 50-60
- 229 Robin PE, Powell DJ, Stansbie JM. Carcinoma of the nasal cavity and paranasal sinuses: incidence and presentation of different histological types. Clin Otolaryngol Allied Sci 1979; 4: 431-456
- 230 Larque AB, Hakim S, Ordi J. et al. High-risk human papillomavirus is transcriptionally active in a subset of sinonasal squamous cell carcinomas. Mod Pathol 2014; 27: 343-351
- 231 Mirghani H, Hartl D, Mortuaire G. et al. Nodal recurrence of sinonasal cancer: Does the risk of cervical relapse justify a prophylactic neck treatment?. Oral Oncol 2013; 49: 374-380
- 232 Castelnau-Marchand P, Levy A, Moya-Plana A. et al. Sinunasales Plattenepithelkarzinom ohne klinische Lymphknotenbeteiligung: Welches Halsmanagement ist das Beste?. Strahlentherapie und Onkol 2016; 192: 537-544
- 233 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian
Papilloma (Malignant Transformation): A Clinicopathologic and
Immunophenotypic Study of 20 Cases Combined with a Comprehensive
Review of the Literature. Head Neck Pathol 2014; 8: 269–286
- 234 Furuta A, Kudo M, Kanai KI. et al. Typical carcinoid tumor arising in the nose and paranasal sinuses – Case report. Auris Nasus Larynx 2010; 37: 381-385
- 235 Guan M, Li Y, Shi ZG. et al. Sarcomatoid carcinoma involving the nasal cavity and paranasal sinus: A rare and highly progressive tumor. Int J Clin Exp Pathol 2014; 7: 4489-4492
- 236 Thompson LDR, Wieneke JA, Miettinen M. et al. Spindle cell (sarcomatoid) carcinomas of the larynx: A clinicopathologic study of 187 cases. Am J Surg Pathol 2002; 26: 153-170
- 237 Lewis JE, Olsen KD, Sebo TJ. Spindle cell carcinoma of the larynx: Review of 26 cases including DNA content and immunohistochemistry. Hum Pathol 1997; 28: 664-673
- 238 Jeng YM, Sung MT, Fang CL. et al. Sinonasal undifferentiated carcinoma and nasopharyngeal-type undifferentiated carcinoma: Two clinically, biologically, and histopathologically distinct entities. Am J Surg Pathol 2002; 26: 371-376
- 239 Leung SuetYi, Yuen SiuTsan, Chung LapPing. et al. Epstein-Barr virus is present in a wide histological spectrum of sinonasal carcinomas. Am J Surg Pathol 1995; 19: 994-1001
- 240
Takakura H, Tachino H, Fujisaka M, Nakajima T, Yamagishi K, Ishida M,
Shojaku H. Lymphoepithelial carcinoma of the maxillary sinus: A case
report and review of the literature. Medicine (Baltimore) 2018;
97(28): e11371. doi:10.1097/MD.0000000000011371. PMID:
29995775; PMCID: PMC6076030
- 241 Frierson HF, Mills SE, Fechner RE. et al. Sinonasal undifferentiated carcinoma. An aggressive neoplasm derived from Schneiderian epithelium and distinct from olfactory neuroblastoma. Am J Surg Pathol 1986; 10: 771-779
- 242 Cerilli LA, Holst VA, Brandwein MS. et al. Sinonasal undifferentiated carcinoma: Immunohistochemical profile and lack of EBV association. Am J Surg Pathol 2001; 25: 156-163
- 243
Abdelmeguid AS, Bell D, Hanna EY. Sinonasal Undifferentiated
Carcinoma. Curr Oncol Rep 2019; 21(3): 26. doi:10.1007/s11912-
019-0776-4. PMID: 30806835
- 244 Musy PY, Reibel JF, Levine PA. Sinonasal undifferentiated carcinoma; The search for a better outcome. Laryngoscope 2002; 112: 1450-1455
- 245 Schröck A, Göke F, van Bremen T. et al. Maligne erkrankungen des sinunasaltrakts. Eine monoinstitutionelle erfahrung von 1996 bis 2010. HNO 2012; 60: 1041-1046
- 246 Rosenthal DI, Barker JL, El-Naggar AK. et al. Sinonasal malignancies with neuroendocrine differentiation: Patterns of failure according to histologic phenotype. Cancer 2004; 101: 2567-2573
- 247 Bell D, Hanna EY. Sinonasal undifferentiated carcinoma: Morphological heterogeneity, diagnosis, management and biological markers. Expert Rev Anticancer Ther 2013; 13: 285-296
- 248 Chambers KJ, Lehmann AE, Remenschneider A. et al. Incidence and survival patterns of sinonasal undifferentiated carcinoma in the United States. J Neurol Surgery, Part B Skull Base 2015; 76: 94-100
- 249 Kuan EC, Arshi A, Mallen-St Clair J, Tajudeen BA, Abemayor E, St John
MA. Significance of Tumor Stage in Sinonasal Undifferentiated
Carcinoma Survival: A Population-Based Analysis. Otolaryngol Head
Neck Surg 2016; 154(4): 667–673. doi:10.1177/01945998
16629649. Epub 2016 Feb 23. PMID: 26908559
- 250 Takahashi Y, Lee J, Pickering C. et al. Human epidermal growth factor receptor 2/neu as a novel therapeutic target in sinonasal undifferentiated carcinoma. Head Neck 2016; 38: E1926-E1934
- 251 Sanchez-Casis G, Devine KD, Weiland LH. Nasal adenocarcinomas that closely simulate colonic carcinomas. Cancer 1971; 28: 714-720
- 252 Barnes L. Intestinal-type adenocarcinoma of the nasal cavity and paranasal sinuses. Am J Surg Pathol 1986; 10: 192-202
- 253 Szablewski V, Solassol J, Poizat F. et al. EGFR expression and KRAS and BRAF mutational status in intestinal-type sinonasal adenocarcinoma. Int J Mol Sci 2013; 14: 5170-5181
- 254 Saber AT, Nielsen LR, Dictor M. et al. K-ras mutations in sinonasal adenocarcinomas in patients occupationally exposed to wood or leather dust. Cancer Lett 1998; 126: 59-65
- 255 Franchi A, Innocenti DRD, Palomba A. et al. Low prevalence of K-RAS, EGF-R and BRAF mutations in sinonasal adenocarcinomas. Implications for anti-EGFR treatments. Pathol Oncol Res 2014; 20: 571-579
- 256 Projetti F, Durand K, Chaunavel A. et al. Epidermal growth factor receptor expression and KRAS and BRAF mutations: Study of 39 sinonasal intestinal-type adenocarcinomas. Hum Pathol 2013; 44: 2116-2125
- 257 Ferrari M, Bossi P, Mattavelli D, Ardighieri L, Nicolai P. Management of
sinonasal adenocarcinomas with anterior skull base extension. J
Neurooncol 2020;150(3): 405–417. doi:10.1007/s11060-019-
03385-8. Epub 2020 Jan 3. PMID: 31897925
- 258 Hoffmann TK, El Hindy N, Müller OM. et al. Vascularised local and free flaps in anterior skull base reconstruction. Eur Arch Oto-Rhino-Laryngology 2013; 270: 899-907
- 259 Hoeben A, van de Winkel L, Hoebers F. et al. Intestinal-type sinonasal adenocarcinomas: The road to molecular diagnosis and personalized treatment. Head Neck 2016; 38: 1564-1570
- 260 Turri-Zanoni M, Battaglia P, Lambertoni A. et al. Treatment strategies for primary early-stage sinonasal adenocarcinoma: A retrospective bi-institutional case-control study. J Surg Oncol 2015; 112: 561-567
- 261 Licitra L, Locati LD, Cavina R. et al. Primary chemotherapy followed by anterior craniofacial resection and radiotherapy for paranasal cancer. Ann Oncol 2003; 14: 367-372
- 262 Jo VY, Mills SE, Cathro HP. et al. Low-grade sinonasal adenocarcinomas: The association with and distinction from respiratory epithelial adenomatoid hamartomas and other glandular lesions. Am J Surg Pathol 2009; 33: 401-408
- 263 Neto AG, Pineda-Daboin K, Luna MA. Sinonasal tract seromucous adenocarcinomas: A report of 12 cases. Ann Diagn Pathol 2003; 7: 154-159
- 264 Heffner DK, Hyams VJ, Hauck KW. et al. Low-grade adenocarcinoma of the nasal cavity and paranasal sinuses. Cancer 1982; 50: 312-322
- 265 Stelow EB, Jo VY, Mills SE. et al. A histologic and immunohistochemical study describing the diversity of tumors classified as sinonasal high-grade nonintestinal adenocarcinomas. Am J Surg Pathol 2011; 35: 971-980
- 266 Bignami M, Lepera D, Volpi L. et al. Sinonasal Non-Intestinal-Type Adenocarcinoma: A Retrospective Review of 22 Patients. World Neurosurg 2018; 120: e962-e969
- 267 Shay A, Ganti A, Raman A. et al. Survival in low-grade and high-grade sinonasal adenocarcinoma: A national cancer database analysis. Laryngoscope 2020; 130: E1-E10
- 268 Shen T, Shi Q, Velosa C. et al. Sinonasal renal cell-like adenocarcinomas: Robust carbonic anhydrase expression. Hum Pathol 2015; 46: 1598-1606
- 269 Heffner DK. Sinonasal and laryngeal salivary gland lesions. In: Surgical Pathology of the Salivary Glands. Philadelphia: WB Saunders; 1991: 544-559
- 270
Akbaba S, Ahmed D, Mock A, Held T, Bahadir S, Lang K, Syed M,
Hoerner-Rieber J, Forster T, Federspil P, Herfarth K, Plinkert P, Debus
J, Adeberg S. Treatment Outcome of 227 Patients with Sinonasal Adenoid
Cystic Carcinoma (ACC) after Intensity Modulated Radiotherapy
and Active Raster-Scanning Carbon Ion Boost: A 10-Year Single-Center
Experience. Cancers (Basel) 2019; 111(11): 1705–417.
doi:10.3390/cancers11111705. PMID: 31683896; PMCID:
PMC6895865
- 271 Spiro RH, Huvos AG, Strong EW. Adenoid cystic carcinoma of salivary origin. A clinicopathologic study of 242 cases. Am J Surg 1974; 128: 512-520
- 272 Spiro RH. Salivary neoplasms: Overview of a 35-year experience with 2,807 patients. Head Neck Surg 1986; 8: 177-184
- 273 Michel G, Joubert M, Delemazure AS. et al. Adenoid cystic carcinoma of the paranasal sinuses: Retrospective series and review of the literature. Eur Ann Otorhinolaryngol Head Neck Dis 2013; 130: 257-262
- 274 Kim GE, Park HC, Keum KC. et al. Adenoid cystic carcinoma of the maxillary antrum. Am J Otolaryngol – Head Neck Med Surg 1999; 20: 77-84
- 275 Chen AM, Bucci MK, Weinberg V. et al. Adenoid cystic carcinoma of the head and neck treated by surgery with or without postoperative radiation therapy: Prognostic features of recurrence. Int J Radiat Oncol Biol Phys 2006; 66: 152-159
- 276 Terhaard CHJ, Lubsen H, Rasch CRN. et al. The role of radiotherapy in the treatment of malignant salivary gland tumors. Int J Radiat Oncol Biol Phys 2005; 61: 103-111
- 277 Garden AS, Weber RS, Ang KK. et al. Postoperative radiation therapy for malignant tumors of minor salivary glands. Outcome and patterns of failure. Cancer 1994; 73: 2563-2569
- 278 Chen AM, Granchi PJ, Garcia J. et al. Local-regional recurrence after surgery without postoperative irradiation for carcinomas of the major salivary glands: Implications for adjuvant therapy. Int J Radiat Oncol Biol Phys 2007; 67: 982-987
- 279 Mendenhall WM, Morris CG, Amdur RJ. et al. Radiotherapy alone or combined with surgery for adenoid cystic carcinoma of the head and neck. Head Neck 2004; 26: 154-162
- 280 Terhaard CHJ, Lubsen H, Van Der Tweel I. et al. Salivary gland carcinoma: Independent prognostic factors for locoregional control, distant metastases, and overall survival: Results of the Dutch Head and Neck Oncology Cooperative Group. Head Neck 2004; 26: 681-693
- 281 Katz TS, Mendenhall WM, Morris CG. et al. Malignant tumors of the nasal cavity and paranasal sinuses. Head Neck 2002; 24: 821-829
- 282 Laramore GE, Krall JM, Griffin TW. et al. Neutron versus photon irradiation for unresectable salivary gland tumors: Final report of an RTOG-MRC randomized clinical trial. Int J Radiat Oncol Biol Phys 1993; 27: 235-240
- 283 Douglas JG, Koh WJ, Austin-Seymour M. et al. Treatment of salivary gland neoplasms with fast neutron radiotherapy. Arch Otolaryngol - Head Neck Surg 2003; 129: 944-948
- 284 Stannard C, Vernimmen F, Carrara H. et al. Malignant salivary gland tumours: Can fast neutron therapy results point the way to carbon ion therapy?. Radiother Oncol 2013; 109: 262-268
- 285 Huber PE, Debus J, Latz D. et al. Radiotherapy for advanced adenoid cystic carcinoma: Neutrons, photons or mixed beam?. Radiother Oncol 2001; 59: 161-167
- 286 Pommier P, Liebsch NJ, Deschler DG. et al. Proton beam radiation therapy for skull base adenoid cystic carcinoma. Arch Otolaryngol - Head Neck Surg 2006; 132: 1242-1249
- 287 Patel NR, Sanghvi S, Khan MN. et al. Demografic trends and disease-specific survival in salivary acinic cell carcinoma: An analysis of 1129 cases. Laryngoscope 2014; 124: 172-178
- 288 Haerle SK, Gullane PJ, Witterick IJ. et al. Sinonasal Carcinomas. Epidemiology, Pathology, and Management. Neurosurg Clin N Am 2013; 24: 39-49
- 289 Neto AG, Pineda-Daboin K, Spencer ML. et al. Sinonasal acinic cell carcinoma: A clinicopathologic study of four cases. Head Neck 2005; 27: 603-607
- 290 Wong A, Leong JL, Ho B. Primary acinic cell carcinoma of the ethmoid sinus. Ear, Nose Throat J 2010; 89
- 291 Biron VL, Lentsch EJ, Gerry DR. et al. Case-control analysis of survival outcomes in sinonasal acinic cell carcinoma. Int Forum Allergy Rhinol 2014; 4: 507-511
- 292 Ellis MA, Graboyes EM, Day TA. et al. Prognostic factors and occult nodal disease in mucoepidermoid carcinoma of the oral cavity and oropharynx: An analysis of the National Cancer Database. Oral Oncol 2017; 72: 174-178
- 293 Auger SR, Patel T, Ganti A. et al. Effect of margin status and pathological grade in treatment of sinonasal mucoepidermoid carcinoma. Laryngoscope. 2020
- 294 Bhattacharyya N. Survival and staging characteristics for non-squamous cell malignancies of the maxillary sinus. Arch Otolaryngol - Head Neck Surg 2003; 129: 334-337
- 295 Wolfish EB, Nelson BL, Thompson LDR. Sinonasal Tract Mucoepidermoid Carcinoma: A Clinicopathologic and Immunophenotypic Study of 19 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2012; 6: 191-207
- 296 Lee YS, Ha SM, Paik SW. et al. Epithelial-myoepithelial carcinoma originating from a minor salivary gland in the nasal septum: A case report and literature review. Medicine (Baltimore) 2020; 99: e19072
- 297 Gore MR. Epithelial-myoepithelial carcinoma: A population-based survival analysis. BMC Ear, Nose Throat Disord 2018; 18
- 298 Morresi-Hauf AT, Reu S, Fertl A. Epithelial-myoepitheliales Karzinom der Trachea: Fallbericht und Literaturübersicht. Pathologe 2013; 34: 56-64
- 299 Roh JL, Chang HR, Choi SH. et al. Clinical utility of 18F-FDG PET for patients with salivary gland malignancies. J Nucl Med 2007; 48: 240-246
- 300 Kim CH, Jeong JS, Kim SR. et al. Endobronchial epithelial-myoepithelial carcinoma of the lung. Thorax 2018; 73: 593-594
- 301 Kim SH, Park SE, Bae HG. et al. Epithelial-myoepithelial carcinoma of the nasopharynx: A case report and review of the literature. Oncol Lett 2015; 10: 927-930
- 302 Fried D, Zanation AM, Huang B. et al. Management of nonesthesioneuroblastoma sinonasal malignancies with neuroendocrine differentiation. Laryngoscope 2012; 122: 2210-2215
- 303 Hong SL, Kim SD, Roh HJ. et al. The sphenoid sinus: An unusual presentation of a typical carcinoid tumor. J Craniofac Surg 2014; 25: e483-e485
- 304 Konukiewitz B, Agaimy A, Weichert W. et al. Neuroendokrine Neoplasien der Kopf-Hals-Region. Pathologe 2018; 39: 27-34
- 305 Ferlito A, Silver CE, Bradford CR. et al. Neuroendocrine neoplasms of the larynx: An overview. Head Neck 2009; 31: 1634-1646
- 306 Lee DH, Cho HH, Cho YB. Typical carcinoid tumor of the nasal cavity. Auris Nasus Larynx 2007; 34: 537-539
- 307 Patel TD, Carniol ET, Vázquez A. et al. Sinonasal fibrosarcoma: Analysis of the Surveillance, Epidemiology, and End Results database. Int Forum Allergy Rhinol 2016; 6: 201-205
- 308 Lartigau E, Lusinchi A, Schwaab G. Sarcomas of nasal cavity and paranasal sinuses: Chondrosarcoma, osteosarcoma and fibrosarcoma. J Laryngol Otol 1994; 108: 947-953
- 309 Koshy M, Rich SE, Mohiuddin MM. Improved Survival With Radiation Therapy in High-Grade Soft Tissue Sarcomas of the Extremities: A SEER Analysis. Int J Radiat Oncol Biol Phys 2010; 77: 203-209
- 310 Wang CP, Chang YL, Ting LL. et al. Malignant fibrous histiocytoma of the sinonasal tract. Head Neck 2009; 31: 85-93
- 311 Szablewski V, Neuville A, Terrier P. et al. Adult sinonasal soft tissue sarcoma: Analysis of 48 cases from the French Sarcoma Group database. Laryngoscope 2015; 125: 615-623
- 312 Gerrand CH, Bell RS, Wunder JS. et al. The influence of anatomic location on outcome in patients with soft tissue sarcoma of the extremity. Cancer 2003; 97: 485-492
- 313 Kwok MMK, Lee S, Hosking P. Leiomyosarcoma: A rare sinonasal malignancy. BMJ Case Rep 2018; 2018
- 314 Tanaka H, Westesson PL, Wilbur DC. Leiomyosarcoma of the maxillary sinus: CT and MRI findings. Br J Radiol 1998; 71: 221-224
- 315 Dobben GD. Leiomyosarcoma of the Nasopharynx. AMA Arch Otolaryngol 1958; 68: 211-213
- 316 Sanghvi S, Misra P, Patel NR. et al. Incidence trends and long-term survival analysis of sinonasal rhabdomyosarcoma. Am J Otolaryngol - Head Neck Med Surg 2013; 34: 682-689
- 317 Wurm J, Constantinidis J, Grabenbauer GG. et al. Rhabdomyosarcomas of the nose and paranasal sinuses: Treatment results in 15 cases. Otolaryngol - Head Neck Surg 2005; 133: 42-50
- 318 Narula AA, Vallis MP, El-Silimy OE. et al. Radiation induced angiosarcomas of the nasopharynx. Eur J Surg Oncol 1986; 12: 147-152
- 319 Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: A study of forty-four cases. Cancer 1981; 48: 1907-1921
- 320 Williamson IG, Ramsden RT. Angiosarcoma of maxillary antrum—association with vinyL chloride exposure. J Laryngol Otol 1988; 102: 464-467
- 321 Nelson BL, Thompson LDR. Sinonasal tract angiosarcoma: A clinicopathologic and immunophenotypic study of 10 cases with a review of the literature. Head Neck Pathol 2007; 1: 1-12
- 322 Fukushima K, Dejima K, Koike S. et al. A case of angiosarcoma of the nasal cavity successfully treated with recombinant interleukin-2. Otolaryngol - Head Neck Surg 2006; 134: 886-887
- 323 Treviño-González JL, Santos-Lartigue R, González-Andrade B. et al. Angiosarcoma of the nasal cavity: A case report. Cases J 2009; 2
- 324 Es-Sbissi F, Nitassi S, Boulaadas M. et al. Sinonasal angiosarcoma. Eur Ann Otorhinolaryngol Head Neck Dis 2015; 132: 161-163
- 325 Agaimy A, Hartmann A. Kopf-Hals-Tumoren: Neues aus der WHO-Klassifikation 2017. Pathologe 2018; 39
- 326 Agaimy A, Haller F, Hartmann A. Sinunasale Tumoren: Neues aus der WHO mit besonderem Fokus auf neue mesenchymale Entitäten. Pathologe 2018; 39: 18-26
- 327 Lewis JT, Oliveira AM, Nascimento AG. et al. Low-grade sinonasal sarcoma with neural and myogenic features: A clinicopathologic analysis of 28 cases. Am J Surg Pathol 2012; 36: 517-525
- 328 Rooper LM, Huang SC, Antonescu CR. et al. Biphenotypic sinonasal sarcoma: An expanded immunoprofile including consistent nuclear β-catenin positivity and absence of SOX10 expression. Hum Pathol 2016; 55: 44-50
- 329 Carter CS, East EG, McHugh JB. Biphenotypic sinonasal sarcoma: A review and update. In: Archives of Pathology and Laboratory Medicine. College of American Pathologists 2018; 1196-1201
- 330 Fritchie KJ, Jin L, Wang X. et al. Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases. Histopathology 2016; 69: 930-936
- 331 Ducatman BS, Scheithauer BW, Piepgras DG. et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57: 2006-2021
- 332 Rodriguez FJ, Folpe AL, Giannini C. et al. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012; 123: 295-319
- 333 James AW, Shurell E, Singh A. et al. Malignant Peripheral Nerve Sheath Tumor. Surg Oncol Clin N Am 2016; 25: 789-802
- 334 Bradtmöller M, Hartmann C, Zietsch J. et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS One 2012; 7: e47595
- 335 Kar M, Suryanarayana Deo SV, Shukla NK. et al. Malignant peripheral nerve sheath tumors (MPNST) - Clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol 2006; 4: 55
- 336 Jo VY, Fletcher CDM. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014; 46: 95-104
- 337 Van De Rijn M, Barr FG, Xiong QBin. et al. Radiation-associated synovial sarcoma. Hum Pathol 1997; 28: 1325-1328
- 338 Deraedt K, Debiec-Rychter M, Sciot R. Radiation-associated synovial sarcoma of the lung following radiotherapy for pulmonary metastasis of Wilms’ tumour [7]. Histopathology 2006; 48: 473-475
- 339 Egger JF, Coindre JM, Benhattar J. et al. Radiation-associated synovial sarcoma: Clinicopathologic and molecular analysis of two cases. Mod Pathol 2002; 15: 998-1004
- 340 Sherman KL, Wayne JD, Chung J. et al. Assessment of multimodality therapy use for extremity sarcoma in the United States. J Surg Oncol 2014; 109: 395-404
- 341 Vlenterie M, Jones RL, Van Der Graaf WTA. Synovial sarcoma diagnosis and management in the era of targeted therapies. Curr Opin Oncol 2015; 27: 316-322
- 342 De Bree E, Zoras O, Hunt JL. et al. Desmoid tumors of the head and neck: A therapeutic challenge. Head Neck 2014; 36: 1517-1526
- 343 Orphanet: Desmoid type fibromatosis. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=8665&Disease_Disease_Search_diseaseType=ORPHA&Disease_Disease_Search_diseaseGroup=873&Disease(s)/groupofdiseases=Desmoid-type-fibromatosis&title=Desmoid-type-fibromatosis&search=Dise
- 344 Colombo C, Foo WC, Whiting D. et al. FAP-related desmoid tumors: A series of 44 patients evaluated in a cancer referral center. Histol Histopathol 2012; 27: 641-649
- 345 Coffin CM, Hornick JL, Zhou H. et al. Gardner fibroma: A clinicopathologic and immunohistochemical analysis of 45 patients with 57 fibromas. Am J Surg Pathol 2007; 31: 410-416
- 346 Emori M, Kaya M, Mitsuhashi T. et al. Desmoid tumor-associated pain is dependent on mast cell expression of cyclooxygenase-2. Diagn Pathol 2014; 9
- 347 Kasper B, Baumgarten C, Garcia J. et al. An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol 2017; 28: 2399-2408
- 348 Bonvalot S, Eldweny H, Haddad V. et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 2008; 34: 462-468
- 349 Penel N, Chibon F, Salas S. Adult desmoid tumors: Biology, management and ongoing trials. Curr Opin Oncol 2017; 29: 268-274
- 350 Huang PW, Tzen CY. Prognostic factors in desmoid-type fibromatosis: A clinicopathological and immunohistochemical analysis of 46 cases. Pathology 2010; 42: 147-150
- 351 Granter SR, Badizadegan K, Fletcher CDM. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: A spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol 1998; 22: 513-525
- 352 Asimakopoulos P, Syed MI, Andrews T. et al. Sinonasal glomangiopericytoma: Is anything new?. Ear, Nose Throat J 2016; 95: E1
- 353 Gengler C, Guillou L. Solitary fibrous tumour and haemangiopericytoma: Evolution of a concept. Histopathology 2006; 48: 63-74
- 354 Thompson LDR, Miettinen M, Wenig BM. Sinonasal-type hemangiopericytoma: A clinicopathologic and immunophenotypic analysis of 104 cases showing perivascular myoid differentiation. Am J Surg Pathol 2003; 27: 737-749
- 355 Catalano PJ, Brandwein M, Shah DK. et al. Sinonasal hemangiopericytomas: A clinicopathologic and immunohistochemical study of seven cases. Head Neck 1996; 18: 42-53
- 356 Billings KR, Fu YS, Calcaterra TC. et al. Hemangiopericytoma of the head and neck. Am J Otolaryngol - Head Neck Med Surg 2000; 21: 238-243
- 357 Carew JF, Singh B, Kraus DH. Hemangiopericytoma of the head and neck. Laryngoscope 1999; 109: 1409-1411
- 358 Weber W, Henkes H, Metz KA. et al. Haemangiopericytoma of the nasal cavity. Neuroradiology 2001; 43: 183-186
- 359 Witkin GB, Rosai J. Solitary fibrous tumor of the upper respiratory tract: A report of six cases. Am J Surg Pathol 1991; 15: 842-848
- 360 Batsakis JG, El-Naggar AK, Hybels RD. Pathology consultation solitary fibrous tumor. Ann Otol Rhinol Laryngol 1993; 102: 74-76
- 361 Künzel J, Hainz M, Ziebart T. et al. Head and neck solitary fibrous tumors: a rare and challenging entity. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1589-1598
- 362 Demicco EG, Park MS, Araujo DM. et al. Solitary fibrous tumor: A clinicopathological study of 110 cases and proposed risk assessment model. Mod Pathol 2012; 25: 1298-1306
- 363 Xue Y, Chai G, Xiao F. et al. Post-operative radiotherapy for the treatment of malignant solitary fibrous tumor of the nasal and paranasal area. Jpn J Clin Oncol 2014; 44: 926-931
- 364 Piccaluga PP, De Falco G, Kustagi M. et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood 2011; 117: 3596-3608
- 365 Chi AC, Weathers DR, Folpe AL. et al. Epithelioid hemangioendothelioma of the oral cavity: Report of two cases and review of the literature. Oral Surgery, Oral Med Oral Pathol Oral Radiol Endodontology 2005; 100: 717-724
- 366 Errani C, Zhang L, Sung YS. et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosom Cancer 2011; 50: 644-653
- 367 Flucke U, Vogels RJC, de Saint Aubain Somerhausen N. et al. Epithelioid Hemangioendothelioma: Clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol 2014; 9
- 368 Bruder E, Alaggio R, Kozakewich HPW. et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol 2012; 15: 26-61
- 369 Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma a vascular tumor often mistaken for a carcinoma. Cancer 1982; 50: 970-981
- 370 Mentzel T, Beham A, Calonje E. et al. Epithelioid hemangioendothelioma of skin and soft tissues: Clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 1997; 21: 363-374
- 371 Deyrup AT, Tighiouart M, Montag AG. et al. Epithelioid hemangioendothelioma of soft tissue: A proposal for risk stratification based on 49 cases. Am J Surg Pathol 2008; 32: 924-927
- 372 Beger M, Antoni C, Haas S. et al. Epitheloides Hämangioendotheliom- drei Patienten, drei Therapieoptionen. Z Gastroenterol 2006; 44: A5_04
- 373 Wong BLK, Lee VNY, Tikka T. et al. Kaposiform haemangioendothelioma of the head and neck. Crit Rev Oncol Hematol 2016; 104: 156-168
- 374 Sun ZJ, Zhang L, Zhang WF. et al. Kaposiform hemangioendothelioma involving the neck. Oral Oncol Extra 2006; 42: 60-65
- 375 Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis 2013; 8: 95
- 376 Pongpruttipan T, Sukpanichnant S, Assanasen T. et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and αβ, γδ, and αβ/γδ T-cell origin: A comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol 2012; 36: 481-499
- 377 Jhuang JY, Chang ST, Weng SF. et al. Extranodal natural killer/T-cell lymphoma, nasal type in Taiwan: A relatively higher frequency of T-cell lineage and poor survival for extranasal tumors. Hum Pathol 2015; 46: 313-321
- 378 De Campos-Lima PO, Gavioli R, Zhang QJ. et al. HLA-A11 epitope loss isolates of Epstein-Barr virus from a highly A11 + population. Science (80-) 1993; 260: 98-100
- 379 Lima PODC, Levitsky V, Brooks J. et al. T cell responses and virus evolution: Loss of hla all-restricted ctl epitopes in epstein-barr virus isolates from highly all-positive populations by selective mutation of anchor residues. J Exp Med 1994; 179: 1297-1305
- 381 Kim SJ, Choi JY, Hyun SH. et al. Risk stratification on the basis of Deauville score on PET-CT and the presence of Epstein-Barr virus DNA after completion of primary treatment for extranodal natural killer/T-cell lymphoma, nasal type: A multicentre, retrospective analysis. Lancet Haematol 2015; 2: e66-e74
- 381 Tse E, Kwong YL. How i treat NK/T-cell lymphomas. Blood 2013; 121: 4997-5005
- 382 Kwong YL, Kim WS, Lim ST. et al. SMILE for natural killer/T-cell lymphoma: Analysis of safety and efficacy from the Asia Lymphoma Study Group. Blood 2012; 120: 2973-2980
- 383 Alexiou C, Kau RJ, Dietzfelbinger H. et al. Extramedullary plasmacytoma: Tumor occurrence and therapeutic concepts. Cancer 1999; 85: 2305-2314
- 384 Miller FR, Lavertu P, Wanamaker JR. et al. Plasmacytomas of the head and neck. Otolaryngol - Head Neck Surg 1998; 119: 614-618
- 385 Patel TD, Vázquez A, Choudhary MM. et al. Sinonasal extramedullary plasmacytoma: A population-based incidence and survival analysis. Int Forum Allergy Rhinol 2015; 5: 862-869
- 386 Soutar R, Lucraft H, Jackson G. et al. Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. Br J Haematol 2004; 124: 717-726
- 387 Lorsbach RB, Hsi ED, Dogan A. et al. Plasma cell myeloma and related neoplasms. In: American Journal of Clinical Pathology 2011; 168-182
- 388 Susnerwala SS, Shanks JH, Banerjee SS. et al. Extramedullary plasmacytoma of the head and neck region: Clinicopathological correlation in 25 cases. Br J Cancer 1997; 75: 921-927
- 389 Hussong JW, Perkins SL, Schnitzer B. et al. Extramedullary plasmacytoma: A form of marginal zone cell lymphoma?. Am J Clin Pathol 1999; 111: 111-116
- 390 Boll M, Parkins E, O’Connor SJM. et al. Extramedullary plasmacytoma are characterized by a „myeloma-like“ immunophenotype and genotype and occult bone marrow involvement. Br J Haematol 2010; 151: 525-527
- 391 Galieni P, Cavo M, Pulsoni A. et al. Clinical outcome of extramedullary plasmacytoma. Haematologica 2000; 85: 47-51
- 392 Sasaki R, Yasuda K, Abe E. et al. Multi-institutional analysis of solitary extramedullary plasmacytoma of the head and neck treated with curative radiotherapy. Int J Radiat Oncol Biol Phys 2012; 82: 626-634
- 393 Knowling MA, Harwood AR, Bergsagel DE. Comparison of extramedullary plasmacytomas with solitary and multiple plasma cell tumors of bone. J Clin Oncol 1983; 1: 255-262
- 394 Hamre M, Hedberg J, Buckley J. et al. Langerhans cell histiocytosis: An exploratory epidemiologic study of 177 cases. Med Pediatr Oncol 1997; 28: 92-97
- 395 Laman JD, Leenen PJM, Annels NE. et al. Langerhans-cell histiocytosis „insight into DC biology“. Trends Immunol 2003; 24: 190-196
- 396 Orphanet: Langerhans Zell Histiozytose. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=389
- 397 Kostyra K, Kostkiewicz B. Langerhans cell histiocytosis of the orbit and frontal sinus of the adult woman: A first case report in Poland. Surg Neurol Int 2019; 10: 234
- 398 Mani S, Thomas R, Mathew J. et al. Langerhan’s cell histiocytosis of sphenoid sinus causing vision loss: A case report. J Nepal Med Assoc 2019; 57: 281-284
- 399 Gündüz K, Palamar M, Parmak N. et al. Eosinophilic granuloma of the orbit: Report of two cases. J AAPOS 2007; 11: 506-508
- 400 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthal Plast Reconstr Surg 2016; 32: 28-34
- 401 Prayer D, Grois N, Prosch H. et al. MR imaging presentation of intracranial disease associated with langerhans cell histiocytosis. Am J Neuroradiol 2004; 25: 880-891
- 402 Allen CE, Li L, Peters TL. et al. Cell-Specific Gene Expression in Langerhans Cell Histiocytosis Lesions Reveals a Distinct Profile Compared with Epidermal Langerhans Cells. J Immunol 2010; 184: 4557-4567
- 403 Hyman DM, Diamond EL, Vibat CRT. et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015; 5: 64-71
- 404 Berres ML, Lim KPH, Peters T. et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014; 211: 669-683
- 405 Wladis EJ, Tomaszewski JE, Gausas RE. Langerhans cell histiocytosis of the orbit 10 years after involvement at other sites. Ophthal Plast Reconstr Surg 2008; 24: 142-143
- 406 Herwig MC, Wojno T, Zhang Q. et al. Langerhans Cell Histiocytosis of the Orbit: Five Clinicopathologic Cases and Review of the Literature. Surv Ophthalmol 2013; 58: 330-340
- 407 Windfuhr JP. Primitive neuroectodermal tumor of the head and neck: Incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 2004; 113: 533-543
- 408 Hafezi S, Seethala RR, Stelow EB. et al. Ewing’s Family of Tumors of the Sinonasal Tract and Maxillary Bone. Head Neck Pathol 2011; 5: 8-16
- 409 Specht K, Sung YS, Zhang L. et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged ewing sarcomas: Further evidence toward distinct pathologic entities. Genes Chromosom Cancer 2014; 53: 622-633
- 410 Lin JK, Liang J. Sinonasal Ewing Sarcoma: A Case Report and Literature Review. Perm J 2018; 22
- 411 Allam A, El-Husseiny G, Khafaga Y. et al. Ewing’s sarcoma of the head and neck: A retrospective analysis of 24 cases. Sarcoma 1999; 3: 11-15
- 412 Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010; 11: 184-192
- 413 Broich G, Pagliari A, Ottaviani F. Esthesioneuroblastoma: A general review of the cases published since the discovery of the tumour in 1924. Anticancer Res 1997; 17: 2683-2706
- 414 Roussel LM, Patron V, Maubert E. et al. New landmarks in endonasal surgery: from nasal bone to anterior cribriform plate including branches of anterior ethmoidal artery and nerve and terminal nerve. Int Forum Allergy Rhinol 2020; 10: 395-404
- 415 Mills SE. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 2002; 15: 264-278
- 416 Thompson LDR. Olfactory Neuroblastoma. Head Neck Pathol 2009; 3: 252-259
- 417 Kadish S, Goodman M, Wang CC. Olfactory neuroblastoma—A clinical analysis of 17 cases. Cancer 1976; 37: 1571-1576
- 418 Morita A, Ebersold MJ, Olsen KD. et al. Esthesioneuroblastoma: Prognosis and management. Neurosurgery 1993; 32: 706-715
- 419 Edge SB, Compton CC. The american joint committee on cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010; 17: 1471-1474
- 420 Norton A. Tumors of the Upper Respiratory Tract and Ear. In: Hyams V, Batsakis J, Michaels L Hrsg. Journal of Clinical Pathology 1989; 335-335
- 421 Mao L, Xia YP, Zhou YN. et al. Activation of sonic hedgehog signaling pathway in olfactory neuroblastoma. Oncology 2009; 77: 231-243
- 422 Ketcham AS, Wilkins RH, Van Buren JM. et al. A combined intracranial facial approach to the paranasal sinuses. Am J Surg 1963; 106: 698-703
- 423 Hanna E, DeMonte F, Ibrahim S. et al. Endoscopic resection of sinonasal cancers with and without craniotomy: Oncologic results. Arch Otolaryngol - Head Neck Surg 2009; 135: 1219-1224
- 424 Su SY, Bell D, Ferrarotto R. et al. Outcomes for olfactory neuroblastoma treated with induction chemotherapy. Head Neck 2017; 39: 1671-1679
- 425 Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: A meta-analysis and review. Lancet Oncol 2001; 2: 683-690
- 426 Ow TJ, Hanna EY, Roberts DB. et al. Optimization of long-term outcomes for patients with esthesioneuroblastoma. Head Neck 2014; 36: 524-530
- 427 Almutuawa DM, Strohl MP, Gruss C. et al. Outcomes of sinonasal mucosal melanomas with endoscopic and open resection: a retrospective cohort study. J Neurooncol. 2020
- 428 Thompson LDR, Wieneke JA, Miettinen M. Sinonasal tract and nasopharyngeal melanomas: A clinicopathologic study of 115 cases with a proposed staging system. Am J Surg Pathol 2003; 27: 594-611
- 429 Moreno MA, Roberts DB, Kupferman ME. et al. Mucosal melanoma of the nose and paranasal sinuses, a contemporary experience from the M. D. Anderson cancer center. Cancer 2010; 116: 2215-2223
- 430 Patel SG, Prasad ML, Escrig M. et al. Primary mucosal malignant melanoma of the head and neck. Head Neck 2002; 24: 247-257
- 431 Zebary A, Jangard M, Omholt K. et al. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: A study of 56 cases. Br J Cancer 2013; 109: 559-564
- 432 Van Essen TH, Van Pelt S, Versluis M. et al. Prognostic parameters in uveal melanoma and their association with bap1 expression. Br J Ophthalmol 2014; 98: 1738-1743
- 433 Dauer EH, Lewis JE, Rohlinger AL. et al. Sinonasal melanoma: A clinicopathologic review of 61 cases. Otolaryngol - Head Neck Surg 2008; 138: 347-352
- 434 Bachar G, Kwok SL, O’Sullivan B. et al. Mucosal melanomas of the head and neck: The Princess Margaret Hospital experience. Head Neck 2008; 30: 1325-1331
- 435 Lund VJ, Chisholm EJ, Howard DJ. et al. Sinonasal malignant melanoma: An analysis of 115 cases assessing outcomes of surgery, postoperative radiotherapy and endoscopic resection. Rhinology 2012; 50: 203-210
- 436 Thierauf J, Glück AM, Plinkert P. et al. Mucosal melanoma of the cranio-facial region: Surgical challenges and therapeutic options. Auris Nasus Larynx 2019; 46: 252-259
- 437 Gore MR, Zanation AM. Survival in sinonasal melanoma: A meta-analysis. J Neurol Surgery, Part B Skull Base 2012; 73: 157-162
- 438 Ganly I, Patel SG, Singh B. et al. Craniofacial resection for malignant melanoma of the skull base: Report of an International Collaborative Study. Arch Otolaryngol - Head Neck Surg 2006; 132: 73-78
- 439 Jarrom D, Paleri V, Kerawala C. et al. Mucosal melanoma of the upper airways tract mucosal melanoma: A systematic review with meta-analyses of treatment. Head Neck 2017; 39: 819-825
- 440 Li J, Kan H, Zhao L. et al. Immune checkpoint inhibitors in advanced or metastatic mucosal melanoma: a systematic review. Ther Adv Med Oncol 2020; 12
- 441 Misra P, Husain Q, Svider PF. et al. Management of sinonasal teratocarcinosarcoma: A systematic review. Am J Otolaryngol - Head Neck Med Surg 2014; 35: 5-11
- 442 Chao KK, Eng TY, Barnes J. et al. Sinonasal Teratocarcinosarcoma. Am J Clin Oncol Cancer Clin Trials 2004; 27: 29-32
- 443 Fukuoka K, Hirokawa M, Shimizu M. et al. Teratocarcinosarcoma of the nasal cavity. Report of a case showing favorable prognosis. Apmis 2000; 108: 553-557
- 444 Heffner DK, Hyams VJ. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses: A clinicopathologic study of 20 cases. Cancer 1984; 53: 2140-2154
- 445 Wei S, Carroll W, Lazenby A. et al. Sinonasal teratocarcinosarcoma: report of a case with review of literature and treatment outcome. Ann Diagn Pathol 2008; 12: 415-425
- 446 Vranic S, Caughron SK, Djuricic S. et al. Hamartomas, teratomas and teratocarcinosarcomas of the head and neck: Report of 3 new cases with clinico-pathologic correlation, cytogenetic analysis, and review of the literature. BMC Ear, Nose Throat Disord 2008; 8
- 447 Lim CCT, Thiagarajan A, Sim CS. et al. Craniospinal dissemination in teratocarcinosarcoma: Case report. J Neurosurg 2008; 109: 321-324
- 448 Terasaka S, Medary MB, Whiting DM. et al. Prolonged survival in a patient with sinonasal teratocarcinosarcoma with cranial extension. Case report. J Neurosurg 1998; 88: 753-756
- 449 Dicke TE, Gates GA. Malignant Teratoma of the Paranasal Sinuses: Report of a Case. Arch Otolaryngol 1970; 91: 391-394
Publication History
Article published online:
30 April 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
Literaturverzeichnis
- 1 Bundesministerium für Gesundheit Seltene Erkrankungen - Bundesgesundheitsministerium. 2019 Im Internet: https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
- 2 Anzahl I. Prävalenzen en und Inzidenzen seltener Krankheiten: Bibliografische Angaben. 2015; 1-52 . Im Internet: https://www.orpha.net/orphacom/cahiers/docs/DE/Pravalenzen_seltener_Krankheiten_Alphabetische_Liste.pdf
- 3 Pedan H, Janosova V, Hajtman A, Calkovsky V. Non-Reflex Defense Mechanisms of Upper Airway Mucosa: Possible Clinical Application. Physiol Res 2020; 69 (Suppl. 01) S55-S67 PMID: 32228012
- 4 Sommer F, Scheithauer M, Kröger R. et al. Niesen als mechanische Abwehr – Numerische Simulation mit Analyse der durchströmten Nasenbereiche. Laryngorhinootologie 2014; 93: 746-750
- 5 Sommer F, Hoffmann TK, Mlynski G. et al. Dreidimensionale Analyse nasaler PhysiologieThree-dimensional analysis of nasal physiology. HNO 2017; 66: 280-289
- 6 Lindemann J, Reichert M, Kröger R. et al. Numerical simulation of humidification and heating during inspiration in nose models with three different located septal perforations. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1795-1800
- 7 Sommer F, Kröger R, Lindemann J. Numerical simulation of humidification and heating during inspiration within an adult nose. Rhinology 2012; 50: 157-164
- 8 Lindemann J, Rettinger G, Kröger R. et al. Numerical simulation of airflow patterns in nose models with differently localized septal perforations. Laryngoscope 2013; 123: 2085-2089
- 9 Götte K, Hörmann K. Sinonasal malignancy: What’s new?. Orl 2004; 66: 85-97
- 10 Barnes L, Eveson JW, Reichart P. et al. World health organization classification of tumours: Pathology and genetics of head and neck tumours. Lyon: International Agency for Research on Cancer; 2005
- 11 Slinger CA, McGarry GW. Nose and sinus tumours: Red flags and referral. Br J Gen Pract 2018; 68: 247-248
- 12 Le QT, Fu KK, Kaplan M. et al. Treatment of maxillary sinus carcinoma: A comparison of the 1997 and 1977 American Joint Committee on Cancer staging systems. Cancer 1999; 86: 1700-1711
- 13 Tiwari R, Hardillo JA, Mehta D. et al. Squamous cell carcinoma of maxillary sinus. Head Neck 2000; 22: 164-169
- 14 Tiwari R, Hardillo JA, Tobi H. et al. Carcinoma of the ethmoid: Results of treatment with conventional surgery and post-operative radiotherapy. Eur J Surg Oncol 1999; 25: 401-405
- 15 Laudien M. Ausgewählte seltene rhinologische Krankheitsbilder Pathogenese – Klinik – Therapie. Laryngorhinootologie 2015; 94: S272-S287
- 16 Hauser LJ, Jensen EL, Mirsky DM. et al. Pediatric anosmia: A case series. Int J Pediatr Otorhinolaryngol 2018; 110: 135-139
- 17 Hummel T, Whitcroft KL, Andrews P. et al. Position paper on olfactory dysfunction. Rhinol J 2017; 0: 1-30
- 18 Orphanet: Anosmie, isolierte kongenitale. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=11807&Disease_Disease_Search_diseaseGroup=anosmie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Anosmie--isolierte-kongenitale&title=Anosmie
isolierte kongenit
- 19 Mahmood AN, Abulaban O, Janjua A. Bilateral olfactory aplasia: Uncommon cause of congenital anosmia. Clin Case Reports 2019; 7: 1456-1457
- 20 Orphanet: Kallmann Syndrom. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=478
- 21 Johnson VP, McMillin JM, Aceto T. et al. A newly recognized neuroectodermal syndrome of familial alopecia, anosmia, deafness, and hypogonadism. Am J Med Genet 1983; 15: 497-506
- 22 Gebril O, Abdelraouf E, Shafie M. et al. Johnson-McMillin microtia syndrome: New additional family. J Fam Med Prim Care 2014; 3: 275
- 23 Orphanet: Arrhinie, isolierte. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=DE&Expert=1134
- 24 Thiele H, Musil A, Nagel F. et al. Familial arhinia, choanal atresia, and microphthalmia. Am J Med Genet 1996; 63: 310-313
- 25 Abukhalaf SA, Zalloum JS, Al Hammouri A. et al. Congenital arrhinia: A case report and literature review. Int J Pediatr Otorhinolaryngol 2020; 135: 110083
- 26 Jung JW, Ha DH, Kim BY. et al. Nasal Reconstruction Using a Customized Three-Dimensional–Printed Stent for Congenital Arhinia: Three-Year Follow-up. Laryngoscope 2019; 129: 582-585
- 27 Núñez-Villaveiran T, Frohner BB, Urcelay PR. et al. Bifid nose – A mild degree of frontonasal dysplasia. A case report. Int J Pediatr Otorhinolaryngol 2013; 77: 1374-1377
- 28 Orphanet: Bifid nose. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2457&Disease_Disease_Search_diseaseGroup=bifid-nose&Disease_Disease_Search_diseaseType=Pat&Disease(s)/groupofdiseases=Bifid-nose&title=Bifid
nose&search=Disease_Search_Simple
- 29 Sedano HO, Michael Cohen M, Jirasek J. et al. Frontonasal dysplasia. J Pediatr 1970; 76: 906-913
- 30 Genç E, Derbent M, Ergin NT. A mild case of frontonasal dysplasia: The rhinologic perspective. Int J Pediatr Otorhinolaryngol 2002; 65: 75-83
- 31 Cohen MM. Perspectives on craniosynostosis: Sutural biology, some well-known syndromes, and some unusual syndromes. J Craniofac Surg 2009; 20: 646-651
- 32 Orphanet: Kraniorhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=de&Expert=157832
- 33 Rüegg EM, Bartoli A, Rilliet B. et al. Management of median and paramedian craniofacial clefts. J Plast Reconstr Aesthetic Surg 2019; 72: 676-684
- 34 Orphanet: Paramedian nasal cleft. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=17067&Disease_Disease_Search_diseaseGroup=paramediannasal-cleft&Disease_Disease_Search_diseasType=Pat&Disease(s)/groupofdiseases=Paramedian-nasal-cleft&title=Paramedian%20nasal%20cleft
- 35 Williams A, Pizzuto M, Brodsky L. et al. Supernumerary nostril: A rare congenital deformity. Int J Pediatr Otorhinolaryngol 1998; 44: 161-167
- 36 Orphanet: Polyrhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17033&Disease_Disease_Search_diseaseGroup=Polyrhinie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Polyrhinie&title=Polyrhinie&search=Disease_Search_Simple
- 37 Orphanet: Nasenlöcher, überzählige. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17034&Disease_Disease_Search_diseaseGroup=Nasenloch-&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Nasenlocher--uberzahlige&title=Nasenl%F6cher,%FCberz%E4hlige
- 38 Gulasi S, Turhan AH, Celik Y. et al. Accessory nostril: A rare congenital nasal anomaly. J Craniofac Surg 2015; 26: e602-e603
- 39 Galiè M, Clauser LC, Tieghi R. et al. The arrhinias: Proboscis lateralis literature review and surgical update. J Cranio-Maxillofacial Surg 2019; 47: 1410-1413
- 40 Harada T, Muraoka M. Proboscis lateralis: A rare bilateral case [7]. Ann Plast Surg 2001; 47: 350-351
- 41 Wolfe SA, Tessier P, Ciminello FS. The Arrhinias. Scand J Plast Reconstr Surg Hand Surg 2009; 43: 177-196
- 42 Rosen Z, Gitlin G. Bilateral Nasal Proboscis: Associated with Unilateral Anophthalmia, Unilateral Diffuse Pigmentation of the Conjunctiva, and Anomalies of the Skull and Brain. AMA Arch Otolaryngol 1959; 70: 545-550
- 43 El-fattah AMA, Naguib A, El-Sisi H. et al. Midline nasofrontal dermoids in children: A review of 29 cases managed at Mansoura University Hospitals. Int J Pediatr Otorhinolaryngol 2016; 83: 88-92
- 44 Orphanet: Nasal dorsum fistula. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=141219
- 45 Jurkov M, Olze H, Klauschen F. et al. IgG4-related orbitopathy as an important differential diagnosis of advanced silent sinus syndrome. HNO 2020; 68: 65-68
- 46 MONTGOMERY WW. Mucocele of the Maxillary Sinus Causing Enophthalmos. Eye Ear Nose Throat Mon 1964; 43: 41-44
- 47 Soparkar CNS, Patrinely JR, Cuaycong MJ. et al. The Silent Sinus Syndrome: A Cause of Spontaneous Enophthalmos. Ophthalmology 1994; 101: 772-778
- 48 Naik RM, Khemani S, Saleh HA. Frontal silent sinus syndrome. Otolaryngol - Head Neck Surg (United States) 2013; 148: 354-355
- 49 McArdle B, Perry C. Ethmoid silent sinus syndrome causing inward displacement of the orbit: Case report. J Laryngol Otol 2010; 124: 206-208
- 50 Wise SK, Wojno TH, DelGaudio JM. Silent sinus syndrome: Lack of orbital findings in early presentation. Am J Rhinol 2007; 21: 489-494
- 51 Brandt MG, Wright ED. The silent sinus syndrome is a form of chronic maxillary atelectasis: A systematic review of all reported cases. Am J Rhinol 2008; 22: 68-73
- 52 Rose GE, Lund VJ. Clinical features and treatment of late enophthalmos after orbital decompression: A condition suggesting cause for idiopathic „imploding antrum“ (silent sinus) syndrome. Ophthalmology 2003; 110: 819-826
- 53 Numa WA, Desai U, Gold DR. et al. Silent sinus syndrome: A case presentation and comprehensive review of all 84 reported cases. Ann Otol Rhinol Laryngol 2005; 114: 688-694
- 54 Davidson JK, Soparkar CNS, Williams JB. et al. Negative sinus pressure and normal predisease imaging in silent sinus syndrome. Arch Ophthalmol 1999; 117: 1653-1654
- 55 Annino DJ, Goguen LA. Silent sinus syndrome. Curr Opin Otolaryngol Head Neck Surg 2008; 16: 22-25
- 56 Hildenbrand T, Klein SB, Schramek N. et al. Rare diseases of the maxillary sinus. HNO 2020; 68: 581-589
- 57 Facon F, Eloy P, Brasseur P. et al. The silent sinus syndrome. Eur Arch Oto-Rhino-Laryngology 2006; 263: 567-571
- 58 Hourany R, Aygun N, Della Santina CC. et al. Silent sinus syndrome: An acquired condition. Am J Neuroradiol 2005; 26: 2390-2392
- 59 Lee DS, Murr AH, Kersten RC. et al. Silent sinus syndrome without opacification of ipsilateral maxillary sinus. Laryngoscope 2018; 128: 2004-2007
- 60 Illner A, Davidson HC, Harnsberger HR. et al. The silent sinus syndrome: Clinical and radiografic findings. Am J Roentgenol 2002; 178: 503-506
- 61 Rose GE, Sandy C, Hallberg L. et al. Clinical and radiologic characteristics of the imploding antrum, or „silent sinus,“ syndrome. Ophthalmology 2003; 110: 811-818
- 62 Weber RK. Aktueller Stand der endonasalen Nasennebenhöhlenchirurgie
[Comprehensive review on endonasal endoscopic sinus
surgery]. Laryngorhinootologie. 2015; 94 Suppl 1; S64–S142.
doi:110.1055/s-0035-1545353. Epub 2015 Apr 10. PMID: 25860497
- 63 Sogg A. Endoscopic sinus surgery. 4th Editio Thieme Verlag; 1993
- 64 Sommer F, Hoffmann T, Lindemann J. et al. Radicality of maxillary sinus surgery and size of the maxillary sinus ostium. HNO 2020; 68: 573-580
- 65 Thomas RD, Graham SM, Carter KD. et al. Management of the orbital floor in silent sinus syndrome. Am J Rhinol 2003; 17: 97-100
- 66 Babar-Craig H, Kayhanian H, de Silva DJ. et al. Spontaneous silent sinus syndrome (Imploding antrum syndrome): Case series of 16 patients. Rhinology 2011; 49: 315-317
- 67 Sivasubramaniam R, Sacks R, Thornton M. Silent sinus syndrome: Dynamic changes in the position of the orbital floor after restoration of normal sinus pressure. J Laryngol Otol 2011; 125: 1239-1243
- 68 Adams WM, Jones RI, Chavda SI. et al. Pneumosinus dilatans: a discussion of four cases and the possible aetiology. Rhinology 1998; 36: 40-402
- 69 Urken ML, Som PM, Lawson W. et al. Abnormally large frontal sinus. II. Nomenclature, pathology, and symptoms. Laryngoscope 1987; 97: 606-611
- 70 Kim WJ, Kim MM. Binasal hemianopia caused by pneumosinus dilatans of the sphenoid sinuses. Indian J Ophthalmol 2019; 67: 1772-1775
- 71 Skolnick CA, Mafee MF, Goodwin JA. Pneumosinus Dilatans of the Sphenoid Sinus Presenting With Visual Loss. J Neuro-ophthalmology 2001; 20: 259-263
- 72 Alatar AA, AlSuliman YA, Alrajhi MS. et al. Maxillary Pneumosinus Dilatans Presenting With Proptosis: A Case Report and Review of the Literature. Clin Med Insights Ear, Nose Throat 2019; 12: 117955061882514
- 73 Sweatman J, Beltechi R. Pneumosinus Dilatans: An exploration into the association between Arachnoid Cyst, Meningioma and the pathogenesis of Pneumosinus Dilatans. Clin Neurol Neurosurg 2019; 185
- 74 Ricci JA. Pneumosinus Dilatans: Over 100 Years Without an Etiology. J Oral Maxillofac Surg 2017; 75: 1519-1526
- 75 Martin AJ, Jarosz JM, Thomas NWM. The strange association of pneumosinus dilatans and arachnoid cyst: Case report and review of the literature. Acta Neurochir (Wien) 2001; 143: 197-201
- 76 Gibbons BA, Miele WR, Florman JE, Heilman CB, Horgan MA
Pneumosinus dilitans and meningioma: a case series and review of
the literature. Neurosurg Focus 2011; 50(5): E13. doi:10.3171/2011.
3.FOCUS1113. PMID: 21529169
- 77 Desai NS, Saboo SS, Khandelwal A. et al. Pneumosinus dilatans: Is it more than an aesthetic concern?. J Craniofac Surg 2014; 25: 418-421
- 78 Draf W, Constantinidis J, Weber R. et al. Pneumosinus Dilatans Frontalis, Atiologie, Symptomatik Und Operationstechnik. Laryngorhinootologie 1996; 75: 660-664
- 79 Cho HS, Hong SJ, Chae HK. et al. Maxillary Sinus Pneumocele Presenting as Aesthetic Deformity: A Case Report With Literature Review. Ear, Nose Throat J 2020; 99: 397-401
- 80 Song M, Ahn SM, Reh DR. et al. Ethmoid pneumocele presenting with exophthalmos 15 years after endoscopic sinus surgery. Allergy Rhinol 2015; 6: 129-132
- 81 Braverman I. Pneumocele of the maxillary sinus with orbital and
trigeminal nerve involvement: case report and review of the
literature. J Otolaryngol Head Neck Surg 2009; 38(2): E35–E38.
PMID: 19442351
- 82 Sasaki T, Yamoto T, Fujita K. et al. A Case of Orbital Emphysema Associated with Frontal Sinus Pneumocele. J Neurol Surg Reports 2013; 74: 054-056
- 83 Bachor E, Weber R, Kahle G. et al. Temporary unilateral amaurosis with pneumosinus dilatans of the sphenoid sinus. Skull Base Surg 1994; 4: 169-175
- 84 Bell AF, Ivan DJ, Munson RA. Barosinus pneumocele: Transient visual loss due to sphenoid sinus pneumocele in a U.S. Air Force pilot. Aviat Sp Environ Med 1995; 66: 276-279
- 85 Choi SJ, Seo ST, Rha KS. et al. Sinonasal organized Hematoma: Clinical features of seventeen cases and a systematic review. Laryngoscope 2015; 125: 2027-2033
- 86 Kim JS, Oh JS, Kwon SH. The increasing incidence of paranasal organizing hematoma: a 20-year experience of 23 cases at a single center. Rhinol J 2016; 54: 176-182
- 87 Pang W, Hu L, Wang H. et al. Organized Hematoma: An Analysis of 84 Cases with Emphasis on Difficult Prediction and Favorable Management. Otolaryngol – Head Neck Surg (United States) 2016; 154: 626-633
- 88 Song HM, Jang YJ, Chung YS. et al. Organizing hematoma of the maxillary sinus. Otolaryngol - Head Neck Surg 2007; 136: 616-620
- 89 Young D. Surgical treatment of male infertility. J Reprod Fertil 1970; 23: 541-542
- 90 Hendry WF, Knight RK, Whitfield HN. et al. Obstructive Azoospermia: Respiratory Function Tests, Electron Microscopy and the Results of Surgery. Br J Urol 1978; 50: 598-604
- 91 Arya AK, Beer HL, Benton J. et al. Does Young’s syndrome exist?. J Laryngol Otol 2009; 123: 477-481
- 92 Hendry WF, Levison DA, Parkinson MC. et al. Testicular obstruction: Clinicopathological studies. Ann R Coll Surg Engl 1990; 72: 396-407
- 93 Shiraishi K, Ono N, Eguchi S. et al. Young’s syndrome associated with situs inversus totalis. Arch Androl 2004; 50: 169-172
- 94 Hasegawa A, Fujita M, Ohe M. et al. A Rare Case of Young’s Syndrome in Japan. Intern Med 1994; 33: 649-653
- 95 Armengot M, Juan G, Carda C. et al. Young’s syndrome: A further cause of chronic rhinosinusitis. Rhinology 1996; 34: 35-37
- 96 Greenstone MA, Rutman A, Hendry WF. et al. Ciliary function in Young’s syndrome. Thorax 1988; 43: 153-154
- 97 Stern BM, Sharma G. Ciliary Dysfunction (Kartagener Syndrome, Primary Ciliary Dyskinesia). StatPearls Publishing; 2019
- 98 Samia H, Khadija B, Agnes H. et al. Long-term outcome of tunisian children with primary ciliary dyskinesia confirmed by transmission electron microscopy. Afr Health Sci 2016; 16: 954-961
- 99 Schofield LM, Duff A, Brennan C. Airway Clearance Techniques for Primary Ciliary Dyskinesia; is the Cystic Fibrosis literature portable?. Paediatr Respir Rev 2018; 25: 73-77
- 100 Alanin MC, Aanaes K, Høiby N. et al. Sinus surgery can improve quality of life, lung infections, and lung function in patients with primary ciliary dyskinesia. Int Forum Allergy Rhinol 2017; 7: 240-247
- 101 Nyilas S, Schlegtendal A, Singer F, Goutaki M, Kuehni CE, Casaulta C,
Latzin P, Koerner-Rettberg C. Alternative inert gas washout outcomes
in patients with primary ciliary dyskinesia. Eur Respir J 2017; 49(1):
1600466. doi:10.1183/13993003.00466-2016. PMID: 28122863
- 102 Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, Dell S,
Eber E, Escudier E, Hirst RA, Hogg C, Jorissen M, Latzin P, Legendre M,
Leigh MW, Midulla F, Nielsen KG, Omran H, Papon JE, Pohunek P,
Redfern B, Rigau D, Rindlisbacher B, Santamaria F, Shoemark A,
Snijders D, Tonia T, Titieni A, Walker WT, Werner C, Bush A, Kuehni
CE. European Respiratory Society guidelines for the diagnosis of
primary ciliary dyskinesia. Eur Respir J 2017; 49(1); 1601090.
doi:10.1183/13993003.00466-2016. PMID: PMC6054534
- 103 Strippoli MPF, Frischer T, Barbato A. et al. Management of primary ciliary dyskinesia in European children: Recommendations and clinical practice. Eur Respir J 2012; 39: 1482-1491
- 104 Barbato A, Frischer T, Kuehni CE. et al. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. In: European Respiratory Journal. Eur Respir J 2009; 1264-1276
- 105 Lucas JS, Burgess A, Mitchison HM. et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child 2014; 99: 850-856
- 106 El-Naggar AK, Chan JKC, Grandis JR et al. WHO Classification of Head
and Neck Tumours. 4th Edition Lyon: International Agency for
Research on Cancer; 2017
- 107 Lombardi D, Tomenzoli D, Buttà L. et al. Limitations and complications of endoscopic surgery for treatment for sinonasal inverted papilloma: A reassessment after 212 cases. Head Neck 2011; 33: 1154-1161
- 108 Stange T, Schultz-Coulon HJ. Zum Chirurgischen Behandlungskonzept bei Invertierten Papillomen der Nase und Nasennebenhöhlen. HNO 2008; 56: 614-622
- 109 Christensen WN, Smith RRL. Schneiderian papillomas. A clinicopathologic study of 67 cases. Hum Pathol 1986; 17: 393-400
- 110 Stammberger H. New aspects in the genesis of inverted papillomas. Laryngol Rhinol Otol (Stuttg) 1983; 62: 249-24955
- 111 Leoncini G, Zanetti L. The papillomas of the sinonasal tract. A comprehensive review. Pathologica 2017; 109: 31-34
- 112 Sarkar FH, Visscher DW, Kintanar EB. et al. Sinonasal Schneiderian papillomas: human papillomavirus typing by polymerase chain reaction. Mod Pathol 1992; 5: 329-332
- 113 Barnes L. Schneiderian papillomas and nonsalivary glandular neoplasms of the head and neck. Mod Pathol 2002; 15: 279-297
- 114 Cunningham MJ, Brantley S, Barnes L. et al. Oncocytic Schneiderian papilloma in a young adult: A rare diagnosis. Otolaryngol Neck Surg 1987; 97: 47-51
- 115 Barnes L, Bedetti C. Oncocytic schneiderian papilloma: A reappraisal of cylindrical cell papilloma of the sinonasal tract. Hum Pathol 1984; 15: 344-351
- 116 Michaels L, Young M. Histogenesis of papillomas of the nose and paranasal sinuses. Arch Pathol Lab Med 1995; 119: 821-826
- 117 Lawson W, Patel ZM. The evolution of management for inverted papilloma: An analysis of 200 cases. Otolaryngol – Head Neck Surg 2009; 140: 330-335
- 118 Lund VJ, Stammberger H, Nicolai P. et al. European position paper on endoscopic management of tumours of the nose, paranasal sinuses and skull base. Rhinol Suppl 2010; 1-143
- 119 Maitra A, Baskin LB, Lee EL. Malignancies arising in oncocytic schneiderian papillomas: A report of 2 cases and review of the literature. Arch Pathol Lab Med 2001; 125: 1365-1367
- 120 Anari S, Carrie S. Sinonasal inverted papilloma: Narrative review. J Laryngol Otol 2010; 124: 705-715
- 121 Attlmayr B, Derbyshire SG, Kasbekar AV et al. Management of
inverted papilloma: Review. In: Journal of Laryngology and Otology.
Cambridge University Press; 2017: 284–289
- 122 Heathcote KJ, Nair SB. The impact of modern techniques on the recurrence rate of inverted papilloma treated by endonasal surgery. Rhinology 2009; 47: 339-344
- 123 Diamantopoulos II, Jones NS, Lowe J. All nasal polyps need histological examination: An audit-based appraisal of clinical practice. J Laryngol Otol 2000; 114: 755-759
- 124 Lloyd GAS, Lloyd GAS. Epithelial Tumours. In: Diagnostic Imaging of the Nose and Paranasal Sinuses 1988; 95-110
- 125 Hildenbrand T, Weber R, Mertens J. et al. Surgery of Inverted Papilloma of the Maxillary Sinus via Translacrimal Approach – Long-Term Outcome and Literature Review. J Clin Med 2019; 8: 1873
- 126 Giotakis E, Knipping S, Kühnel T. et al. Unilateral diseases of the maxillary sinus. HNO 2020; 68: 623-636
- 127 Vrabec DP. The inverted schneiderian papilloma: A 25-year study. Laryngoscope 1994; 104: 582-605
- 128 Bielamowicz S, Calcaterra TC, Watson D. Inverting papilloma of the head and neck: The UCLA update. Otolaryngol – Head Neck Surg 1993; 109: 71-76
- 129 Kraft M, Simmen D, Kaufmann T. et al. Long-term results of endonasal sinus surgery in sinonasal papillomas. Laryngoscope 2003; 113: 1541-1547
- 130 Tanna N, Edwards JD, Aghdam H. et al. Transnasal endoscopic medial maxillectomy as the initial oncologic approach to sinonasal neoplasms: The anatomic basis. Arch Otolaryngol – Head Neck Surg 2007; 133: 1139-1142
- 131 Busquets JM, Hwang PH. Endoscopic resection of sinonasal inverted papilloma: A meta-analysis. Otolaryngol – Head Neck Surg 2006; 134: 476-482
- 132 Veeresh M, Sudhakara M, Girish G. et al. Leiomyoma: A rare tumor in the head and neck and oral cavity: Report of 3 cases with review. J Oral Maxillofac Pathol 2013; 17: 281-287
- 133 Agaimy A, Michal M, Thompson LDR. et al. Angioleiomyoma of the Sinonasal Tract: Analysis of 16 Cases and Review of the Literature. Head Neck Pathol 2015; 9: 463-473
- 134 Heyman JNS, Jones LM, Hilton JM. et al. Angioleiomyoma of the inferior turbinate: A rare cause of isolated facial pain. Ann R Coll Surg Engl 2020; 102: E20-E22
- 135 Lau YW, Tharumalingam V, Tan TY. et al. Sinonasal angioleiomyoma. Med J Malaysia 2016; 71: 154-155
- 136 Mathieu T, Verbruggen A, Goovaerts G. et al. Vascular leiomyoma of the nasal cavity: case report and literature review. Eur Arch Oto-Rhino-Laryngology 2017; 274: 579-583
- 137 Poncet A, Dor L. Botryomycose humaine. Rev Chir 1897; 18: 996-1003
- 138 Hartzell M. Granuloma pyogenicum (botryomycosis of French authors). J Cutan Dis 1904; 22: 520-523
- 139 Mills SE, Cooper PH, Fechner RE. Lobular capillary hemangioma: The underlying lesion of pyogenic granuloma. A study of 73 cases from the oral and nasal mucous membranes. Am J Surg Pathol 1980; 4: 471-479
- 140 Fu Y-S, Perzin KH. Nonepithelial tumors of the nasal cavity, paranasal sinuses, and nasopharynx. A clinicopathologic study VI. Fibrous tissue tumors (fibroma, fibromatosis, fibrosarcoma). Cancer 1976; 37: 2912-2928
- 141 Thompson LDR, Fanburg-Smith JC. Update on Select Benign Mesenchymal and Meningothelial Sinonasal Tract Lesions. Head Neck Pathol 2016; 10: 95-108
- 142 Sammut SJ, Tomson N, Corrie P. Pyogenic granuloma as a cutaneous adverse effect of vemurafenib. N Engl J Med 2014; 371: 1265-1267
- 143 Smith SC, Patel RM, Lucas DR. et al. Sinonasal Lobular Capillary Hemangioma: A Clinicopathologic Study of 34 Cases Characterizing Potential for Local Recurrence. Head Neck Pathol 2013; 7: 129-134
- 144 Sunaryo PL, Svider PF, Husain Q. et al. Schwannomas of the sinonasal tract and anterior skull base: A systematic review of 94 cases. Am J Rhinol Allergy 2014; 28: 39-49
- 145 Shugar JMA, Son PM, Biller HF. et al. Peripheral nerve sheath tumors of the paranasal sinuses. Head Neck Surg 1981; 4: 72-76
- 146 Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg 1994; 81: 362-373
- 147 Ansari I, Ansari A, Graison AA. et al. Head and Neck Schwannomas: A Surgical Challenge – A Series of 5 Cases. Case Rep Otolaryngol 2018; 2018: 1-10
- 148 Azani AB, Bishop JA, Thompson LDR. Sinonasal Tract Neurofibroma: A Clinicopathologic Series of 12 Cases with a Review of the Literature. Head Neck Pathol 2015; 9: 323-333
- 149 Annino DJ, Domanowski GF, Vaughan CW. A Rare Cause of Nasal Obstruction: A Solitary Neurofibroma. Otolaryngol Neck Surg 1991; 104: 484-488
- 150 Perzin KH, Panyu H, Wechter S. Nonepithelial tumors of the nasal cavity, paranasal sinuses and nasopharynx. A clinicopathologic study. XII: Schwann cell tumors (neurilemoma, neurofibroma, malignant schwannoma). Cancer 1982; 50: 2193-2202
- 151 Friedman CD, Costantino PD, Teitelbaum B. et al. Primary extracranial meningiomas of the head and neck. Laryngoscope 1990; 100: 41-48
- 152 Thompson LDR, Gyure KA. Extracranial sinonasal tract meningiomas: A clinicopathologic study of 30 cases with a review of the literature. Am J Surg Pathol 2000; 24: 640-650
- 153 Ho K-L. Primary meningioma of the nasal cavity and paranasal sinuses. Cancer 1980; 46: 1442-1447
- 154 Perry A, Scheithauer BW, Stafford SL. et al. „Malignancy“ in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer 1999; 85: 2046-2056
- 155 Perry A, Stafford SL, Scheithauer BW. et al. Meningioma grading: An analysis of histologic parameters. Am J Surg Pathol 1997; 21: 1455-1465
- 156 Rushing EJ, Bouffard JP, McCall S. et al. Primary extracranial meningiomas: An analysis of 146 cases. Head Neck Pathol 2009; 3: 116-130
- 157 Mnejja M, Hammami B, Bougacha L. et al. Primary sinonasal meningioma. Eur Ann Otorhinolaryngol Head Neck Dis 2012; 129: 47-50
- 158 Wolf A, Safran B, Pock J. et al. Surgical treatment of paranasal sinus osteomas: A single center experience of 58 cases. Laryngoscope. 2019
- 159 Atallah N, Jay MM. Osteomas of the paranasal sinuses. J Laryngol Otol 1981; 95: 291-304
- 160 Furlaneto EC, Rocha JRM, Heitz C. Osteoma of the zygomatic arch - Report of a case. Int J Oral Maxillofac Surg 2004; 33: 310-311
- 161 Summers LE, Mascott CR, Tompkins JR. et al. Frontal sinus osteoma associated with cerebral abscess formation: A case report. Surg Neurol 2001; 55: 235-239
- 162 Eller R, Sillers M. Common Fibro-osseous Lesions of the Paranasal Sinuses. Otolaryngol Clin North Am 2006; 39: 585-600
- 163 Schick B, Steigerwald C, El Tahan AER. et al. The role of endonasal surgery in the management of frontoethmoidal osteomas. Rhinology 2001; 39: 66-70
- 164 Schick B, Dlugaiczyk J. Benign tumors of the nasal cavity and paranasal sinuses. In: Rhinology and Facial Plastic Surgery 2009; 377-385
- 165 Buyuklu F, Akdogan MV, Ozer C. et al. Growth characteristics and clinical manifestations of the paranasal sinus osteomas. Otolaryngol - Head Neck Surg 2011; 145: 319-323
- 166 Golant A, Zeichner JA. Gardner Syndrome. StatPearls Publishing; 2014
- 167 Pinto RS, Simons A, Verma R. et al. Gardener-associated fibroma: An unusual cause of upper airway obstruction. BMJ Case Rep 2018; 2018
- 168 Yang A, Sisson R, Gupta A, Tiao G, Geller JI. Germline APC mutations
in hepatoblastoma. Pediatr Blood Cancer 2018; 65(4); doi:10.1002/
pbc.26892. Epub 2017 Dec 18. PMID: 29251405
- 169 Kiessling P, Dowling E, Huang Y, Ho ML, Balakrishnan K, Weigel BJ,
Highsmith WE Jr, Niu Z, Schimmenti LA. Identification of aggressive
Gardner syndrome phenotype associated with a de novo APC variant,
c.4666dup. Cold Spring Harb Mol Case Stud 2019; 5(2): a003640.
doi:10.1101/mcs.a003640. PMID: 30696621; PMCID: PMC6549566
- 170 Burke AB, Collins MT, Boyce AM. Fibrous dysplasia of bone: craniofacial and dental implications. Oral Dis 2017; 23: 697-708
- 171 Erkmen K, Al-Mefty O, Adada B. Tumors of the skull base. In: Oncology of CNS Tumors Philadelphia: WB Saunders; 2010: 279-307
- 172 Heller AJ, DiNardo LJ, Massey D. Fibrous dysplasia, chondrosarcoma, and McCune-Albright syndrome. Am J Otolaryngol – Head Neck Med Surg 2001; 22: 297-301
- 173 Kos M, Luczak K, Godzinski J. et al. Treatment of monostotic fibrous dysplasia with pamidronate. J Cranio-Maxillofacial Surg 2004; 32: 10-15
- 174 Collins MT, Kushner H, Reynolds JC. et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res 2005; 20: 219-226
- 175 Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: Age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int 2008; 19: 57-63
- 176 Devogelaer JP. New uses of bisphosphonates: Osteogenesis imperfecta. Curr Opin Pharmacol 2002; 2: 748-753
- 177 Schreiber A, Villaret AB, Maroldi R. et al. Fibrous dysplasia of the sinonasal tract and adjacent skull base. Curr Opin Otolaryngol Head Neck Surg 2012; 20: 45-52
- 178 Amit M, Collins MT, FitzGibbon EJ. et al. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia – a meta-analysis. PLoS One 2011; 6
- 179 McCune D. Osteitis fibrosa cystica: The case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 1936; 52: 743-744
- 180 Leet AI, Chebli C, Kushner H. et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res 2004; 19: 571-577
- 181 Boyce AM, Brewer C, Deklotz TR. et al. Association of hearing loss and otologic outcomes with fibrous dysplasia. JAMA Otolaryngol – Head Neck Surg 2018; 144: 102-107
- 182 Deklotz TR, Kim HJ, Kelly M. et al. Sinonasal disease in polyostotic fibrous dysplasia and McCune-Albright Syndrome. Laryngoscope 2013; 123: 823-828
- 183 Boyce AM, Glover M, Kelly MH. et al. Optic neuropathy in McCune-Albright syndrome: Effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab 2013; 98
- 184 Plotkin H, Rauch F, Zeitlin L. et al. Effect of Pamidronate Treatment in Children with Polyostotic Fibrous Dysplasia of Bone. J Clin Endocrinol Metab 2003; 88: 4569-4575
- 185 DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev 2007; 4: 440-445
- 186 Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; (03) 12 PMID: 18489744 PMCID: PMC2459161
- 187 Feldman MD, Kelly M, Rao VM. et al. Fibrous dysplasia of the paranasal sinuses. Otolaryngol Neck Surg 1986; 95: 222-225
- 188 Ikeda K, Suzuki H, Oshima T. et al. Endonasal endoscopic management in fibrous dysplasia of the paranasal sinuses. Am J Otolaryngol – Head Neck Med Surg 1997; 18: 415-418
- 189 Uzun C, Adali MK, Koten M. et al. McCune-Albright syndrome with fibrous dysplasia of the paranasal sinuses. Rhinology 1999; 37: 122-124
- 190 Lee JS, Fitzgibbon E, Butman JA. et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 2002; 347: 1670-1676
- 191 Wenig BM, Heffner DK. Respiratory epithelial adenomatoid hamartomas of the sinonasal tract and nasopharynx: A clinicopathologic study of 31 cases. Ann Otol Rhinol Laryngol 1995; 104: 639-645
- 192 Delbrouck C, Fernandez Aguilar S, Choufani G. et al. Respiratory epithelial adenomatoid hamartoma associated with nasal polyposis. Am J Otolaryngol – Head Neck Med Surg 2004; 25: 282-284
- 193 Endo R, Matsuda H, Takahashi M. et al. Respiratory epithelial adenomatoid hamartoma in the nasal cavity. Acta Otolaryngol 2002; 122: 398-400
- 194 Ingram WF, Noone MC, Gillespie MB. Respiratory epithelial adenomatoid hamartoma: A case report. Ear, Nose Throat J 2006; 85: 190-192
- 195 Athre R, Ducic Y. Frontal sinus hamartomas. Am J Otolaryngol – Head Neck Med Surg 2005; 26: 419-421
- 196 Flavin R, Russell J, Phelan E. et al. Chondro-osseous respiratory epithelial adenomatoid hamartoma of the nasal cavity: A case report. Int J Pediatr Otorhinolaryngol 2005; 69: 87-91
- 197 Fitzhugh VA, Mirani N. Respiratory epithelial adenomatoid hamartoma: A review. Head Neck Pathol 2008; 2: 203-208
- 198 Braun H, Beham A, Stammberger H. Respiratorisches epitheliales adenomatoides hamartom der nasenhaupthöhle: Fallbericht und literaturübersicht. Laryngorhinootologie 2003; 82: 416-420
- 199 Himi Y, Yoshizaki T, Sato K. et al. Respiratory epithelial adenomatoid hamartoma of the maxillary sinus. J Laryngol Otol 2002; 116: 317-318
- 200 Safi C, Li C, Tabaee A. et al. Outcomes and imaging findings of respiratory epithelial adenomatoid hamartoma: a systematic review. Int Forum Allergy Rhinol 2019; 9: 674-680
- 201 Lima NB, Jankowski R, Georgel T. et al. Respiratory adenomatoid hamartoma must be suspected on CT-scan enlargement of the olfactory clefts. Rhinology 2006; 44: 264-269
- 202 Hawley KA, Ahmed M, Sindwani R. CT findings of sinonasal respiratory epithelial adenomatoid hamartoma: A closer look at the olfactory clefts. Am J Neuroradiol 2013; 34: 1086-1090
- 203 Metselaar RM, Stel HV, Van Der Baan S. Respiratory epithelial adenomatoid hamartoma in the nasopharynx. J Laryngol Otol 2005; 119: 476-478
- 204 Hawley KA, Pabon S, Hoschar AP. et al. The presentation and clinical significance of sinonasal respiratory epithelial adenomatoid hamartoma (REAH). Int Forum Allergy Rhinol 2013; 3: 248-253
- 205 Schafer DR, Thompson LDR, Smith BC. et al. Primary ameloblastoma of the sinonasal tract: A clinicopathologic study of 24 cases. Cancer 1998; 82: 667-674
- 206 Shahidi S, Bronoosh P, Daneshbod Y. Follicular ameloblastoma presenting as a sinonasal tumor. Iran Red Crescent Med J 2012; 14: 113-116
- 207 Hertog D, van der Waal I. Ameloblastoma of the jaws: A critical reappraisal based on a 40-years single institution experience. Oral Oncol 2010; 46: 61-64
- 208 Barrena BG, Phillips BJ, Moran KJ. et al. Sinonasal Ameloblastoma. Head Neck Pathol 2019; 13: 247-250
- 209
Golbin DA, Ektova AP, Demin MO, Lasunin N, Cherekaev VA. Nasal
Chondromesenchymal Hamartoma with Skull Base and Orbital
Involvement: Case Presentation. Cureus 2018; 10(6): e2892.
doi:10.7759/cureus.2892. PMID: 30167348; PMCID: PMC6112910
- 210 McDermott MB, Ponder TB, Dehner LP. Nasal chondromesenchymal hamartoma: An upper respiratory tract analogue of the chest wall mesenchymal hamartoma. Am J Surg Pathol 1998; 22: 425-433
- 211 Ozolek JA, Carrau R, Barnes EL. et al. Nasal chondromesenchymal hamartoma in older children and adults: Series and immunohistochemical analysis. Arch Pathol Lab Med 2005; 129: 1444-1450
- 212 Chan TY. World Health Organization classification of tumours: Pathology & genetics of tumours of the urinary system and male genital organs. Urology 2005; 65: 214-215
- 213 Seibert RW, Seibert JJ, Jimenez JF. et al. Nasopharyngeal brain heterotopia – a cause of upper airway obstruction in infancy. Laryngoscope 1984; 94: 818-819
- 214 Kanjanawasee D, Chaowanapanja P, Keelawat S. et al. Sphenoid Sinus Cholesteatoma – Complications and Skull Base Osteomyelitis: Case Report and Review of Literature. Clin Med Insights Case Reports 2019; 12
- 215 Hansen S, Sørensen CH, Stage J. et al. Massive cholesteatoma of the frontal sinus: Case report and review of the literature. Auris Nasus Larynx 2007; 34: 387-392
- 216
Gey A, Plontke SK, Scheller C, Kösling S, Fathke C, Glien A. Seltene
Diagnose einer knochendestruierenden Läsion der Keilbeinhöhle
[Rare diagnosis of a bone-destructive lesion of the sphenoid sinus].
HNO. 2020 Dec 14. German. doi:10.1007/s00106-020-00974-2.
Epub ahead of print. PMID: 33315128
- 217 Ansa B, Goodman M, Ward K. et al. Paranasal sinus squamous cell carcinoma incidence and survival based on surveillance, epidemiology, and end results data, 1973 to 2009. Cancer 2013; 119: 2602-2610
- 218 Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: A historical analysis of population-based data. Head Neck 2012; 34: 877-885
- 219 Sanghvi S, Khan MN, Patel NR. et al. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4994 patients. Laryngoscope 2014; 124: 76-83
- 220 Youlden DR, Cramb SM, Peters S. et al. International comparisons of the incidence and mortality of sinonasal cancer. Cancer Epidemiol 2013; 37: 770-779
- 221 Hayes RB, Kardaun JWPF, De Bruyn A. Tobacco use and sinonasal cancer: A case-control study. Br J Cancer 1987; 56: 843-846
- 222 Brinton LA, Blot WJ, Becker JA. et al. A case-control study of cancers of the nasal cavity and paranasal sinuses. Am J Epidemiol 1984; 119: 896-906
- 223 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian Papilloma (Malignant Transformation): A Clinicopathologic and Immunophenotypic Study of 20 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2014; 8: 269-286
- 224 Dulguerov P, Jacobsen M, Allal A. et al. Nasal and paranasal sinus carcinoma: are we making progress? A series of 220 patients and a systematic review. Cancer 2001; 92: 3012-3029
- 225 Thorup C, Sebbesen L, Danø H. et al. Carcinoma of the nasal cavity and paranasal sinuses in Denmark 1995-2004. Acta Oncol (Madr) 2010; 49: 389-394
- 226 El-Mofty SK, Lu DW. Prevalence of high-risk human papillomavirus DNA in nonkeratinizing (cylindrical cell) carcinoma of the sinonasal tract: A distinct clinicopathologic and molecular disease entity. Am J Surg Pathol 2005; 29: 1367-1372
- 227 Bishop JA, Guo TW, Smith DF. et al. Human papillomavirus-related carcinomas of the sinonasal tract. Am J Surg Pathol 2013; 37: 185-192
- 228 Osborn DA. Nature and behavior of transitional tumors in the upper respiratory tract. Cancer 1970; 25: 50-60
- 229 Robin PE, Powell DJ, Stansbie JM. Carcinoma of the nasal cavity and paranasal sinuses: incidence and presentation of different histological types. Clin Otolaryngol Allied Sci 1979; 4: 431-456
- 230 Larque AB, Hakim S, Ordi J. et al. High-risk human papillomavirus is transcriptionally active in a subset of sinonasal squamous cell carcinomas. Mod Pathol 2014; 27: 343-351
- 231 Mirghani H, Hartl D, Mortuaire G. et al. Nodal recurrence of sinonasal cancer: Does the risk of cervical relapse justify a prophylactic neck treatment?. Oral Oncol 2013; 49: 374-380
- 232 Castelnau-Marchand P, Levy A, Moya-Plana A. et al. Sinunasales Plattenepithelkarzinom ohne klinische Lymphknotenbeteiligung: Welches Halsmanagement ist das Beste?. Strahlentherapie und Onkol 2016; 192: 537-544
- 233 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian
Papilloma (Malignant Transformation): A Clinicopathologic and
Immunophenotypic Study of 20 Cases Combined with a Comprehensive
Review of the Literature. Head Neck Pathol 2014; 8: 269–286
- 234 Furuta A, Kudo M, Kanai KI. et al. Typical carcinoid tumor arising in the nose and paranasal sinuses – Case report. Auris Nasus Larynx 2010; 37: 381-385
- 235 Guan M, Li Y, Shi ZG. et al. Sarcomatoid carcinoma involving the nasal cavity and paranasal sinus: A rare and highly progressive tumor. Int J Clin Exp Pathol 2014; 7: 4489-4492
- 236 Thompson LDR, Wieneke JA, Miettinen M. et al. Spindle cell (sarcomatoid) carcinomas of the larynx: A clinicopathologic study of 187 cases. Am J Surg Pathol 2002; 26: 153-170
- 237 Lewis JE, Olsen KD, Sebo TJ. Spindle cell carcinoma of the larynx: Review of 26 cases including DNA content and immunohistochemistry. Hum Pathol 1997; 28: 664-673
- 238 Jeng YM, Sung MT, Fang CL. et al. Sinonasal undifferentiated carcinoma and nasopharyngeal-type undifferentiated carcinoma: Two clinically, biologically, and histopathologically distinct entities. Am J Surg Pathol 2002; 26: 371-376
- 239 Leung SuetYi, Yuen SiuTsan, Chung LapPing. et al. Epstein-Barr virus is present in a wide histological spectrum of sinonasal carcinomas. Am J Surg Pathol 1995; 19: 994-1001
- 240
Takakura H, Tachino H, Fujisaka M, Nakajima T, Yamagishi K, Ishida M,
Shojaku H. Lymphoepithelial carcinoma of the maxillary sinus: A case
report and review of the literature. Medicine (Baltimore) 2018;
97(28): e11371. doi:10.1097/MD.0000000000011371. PMID:
29995775; PMCID: PMC6076030
- 241 Frierson HF, Mills SE, Fechner RE. et al. Sinonasal undifferentiated carcinoma. An aggressive neoplasm derived from Schneiderian epithelium and distinct from olfactory neuroblastoma. Am J Surg Pathol 1986; 10: 771-779
- 242 Cerilli LA, Holst VA, Brandwein MS. et al. Sinonasal undifferentiated carcinoma: Immunohistochemical profile and lack of EBV association. Am J Surg Pathol 2001; 25: 156-163
- 243
Abdelmeguid AS, Bell D, Hanna EY. Sinonasal Undifferentiated
Carcinoma. Curr Oncol Rep 2019; 21(3): 26. doi:10.1007/s11912-
019-0776-4. PMID: 30806835
- 244 Musy PY, Reibel JF, Levine PA. Sinonasal undifferentiated carcinoma; The search for a better outcome. Laryngoscope 2002; 112: 1450-1455
- 245 Schröck A, Göke F, van Bremen T. et al. Maligne erkrankungen des sinunasaltrakts. Eine monoinstitutionelle erfahrung von 1996 bis 2010. HNO 2012; 60: 1041-1046
- 246 Rosenthal DI, Barker JL, El-Naggar AK. et al. Sinonasal malignancies with neuroendocrine differentiation: Patterns of failure according to histologic phenotype. Cancer 2004; 101: 2567-2573
- 247 Bell D, Hanna EY. Sinonasal undifferentiated carcinoma: Morphological heterogeneity, diagnosis, management and biological markers. Expert Rev Anticancer Ther 2013; 13: 285-296
- 248 Chambers KJ, Lehmann AE, Remenschneider A. et al. Incidence and survival patterns of sinonasal undifferentiated carcinoma in the United States. J Neurol Surgery, Part B Skull Base 2015; 76: 94-100
- 249 Kuan EC, Arshi A, Mallen-St Clair J, Tajudeen BA, Abemayor E, St John
MA. Significance of Tumor Stage in Sinonasal Undifferentiated
Carcinoma Survival: A Population-Based Analysis. Otolaryngol Head
Neck Surg 2016; 154(4): 667–673. doi:10.1177/01945998
16629649. Epub 2016 Feb 23. PMID: 26908559
- 250 Takahashi Y, Lee J, Pickering C. et al. Human epidermal growth factor receptor 2/neu as a novel therapeutic target in sinonasal undifferentiated carcinoma. Head Neck 2016; 38: E1926-E1934
- 251 Sanchez-Casis G, Devine KD, Weiland LH. Nasal adenocarcinomas that closely simulate colonic carcinomas. Cancer 1971; 28: 714-720
- 252 Barnes L. Intestinal-type adenocarcinoma of the nasal cavity and paranasal sinuses. Am J Surg Pathol 1986; 10: 192-202
- 253 Szablewski V, Solassol J, Poizat F. et al. EGFR expression and KRAS and BRAF mutational status in intestinal-type sinonasal adenocarcinoma. Int J Mol Sci 2013; 14: 5170-5181
- 254 Saber AT, Nielsen LR, Dictor M. et al. K-ras mutations in sinonasal adenocarcinomas in patients occupationally exposed to wood or leather dust. Cancer Lett 1998; 126: 59-65
- 255 Franchi A, Innocenti DRD, Palomba A. et al. Low prevalence of K-RAS, EGF-R and BRAF mutations in sinonasal adenocarcinomas. Implications for anti-EGFR treatments. Pathol Oncol Res 2014; 20: 571-579
- 256 Projetti F, Durand K, Chaunavel A. et al. Epidermal growth factor receptor expression and KRAS and BRAF mutations: Study of 39 sinonasal intestinal-type adenocarcinomas. Hum Pathol 2013; 44: 2116-2125
- 257 Ferrari M, Bossi P, Mattavelli D, Ardighieri L, Nicolai P. Management of
sinonasal adenocarcinomas with anterior skull base extension. J
Neurooncol 2020;150(3): 405–417. doi:10.1007/s11060-019-
03385-8. Epub 2020 Jan 3. PMID: 31897925
- 258 Hoffmann TK, El Hindy N, Müller OM. et al. Vascularised local and free flaps in anterior skull base reconstruction. Eur Arch Oto-Rhino-Laryngology 2013; 270: 899-907
- 259 Hoeben A, van de Winkel L, Hoebers F. et al. Intestinal-type sinonasal adenocarcinomas: The road to molecular diagnosis and personalized treatment. Head Neck 2016; 38: 1564-1570
- 260 Turri-Zanoni M, Battaglia P, Lambertoni A. et al. Treatment strategies for primary early-stage sinonasal adenocarcinoma: A retrospective bi-institutional case-control study. J Surg Oncol 2015; 112: 561-567
- 261 Licitra L, Locati LD, Cavina R. et al. Primary chemotherapy followed by anterior craniofacial resection and radiotherapy for paranasal cancer. Ann Oncol 2003; 14: 367-372
- 262 Jo VY, Mills SE, Cathro HP. et al. Low-grade sinonasal adenocarcinomas: The association with and distinction from respiratory epithelial adenomatoid hamartomas and other glandular lesions. Am J Surg Pathol 2009; 33: 401-408
- 263 Neto AG, Pineda-Daboin K, Luna MA. Sinonasal tract seromucous adenocarcinomas: A report of 12 cases. Ann Diagn Pathol 2003; 7: 154-159
- 264 Heffner DK, Hyams VJ, Hauck KW. et al. Low-grade adenocarcinoma of the nasal cavity and paranasal sinuses. Cancer 1982; 50: 312-322
- 265 Stelow EB, Jo VY, Mills SE. et al. A histologic and immunohistochemical study describing the diversity of tumors classified as sinonasal high-grade nonintestinal adenocarcinomas. Am J Surg Pathol 2011; 35: 971-980
- 266 Bignami M, Lepera D, Volpi L. et al. Sinonasal Non-Intestinal-Type Adenocarcinoma: A Retrospective Review of 22 Patients. World Neurosurg 2018; 120: e962-e969
- 267 Shay A, Ganti A, Raman A. et al. Survival in low-grade and high-grade sinonasal adenocarcinoma: A national cancer database analysis. Laryngoscope 2020; 130: E1-E10
- 268 Shen T, Shi Q, Velosa C. et al. Sinonasal renal cell-like adenocarcinomas: Robust carbonic anhydrase expression. Hum Pathol 2015; 46: 1598-1606
- 269 Heffner DK. Sinonasal and laryngeal salivary gland lesions. In: Surgical Pathology of the Salivary Glands. Philadelphia: WB Saunders; 1991: 544-559
- 270
Akbaba S, Ahmed D, Mock A, Held T, Bahadir S, Lang K, Syed M,
Hoerner-Rieber J, Forster T, Federspil P, Herfarth K, Plinkert P, Debus
J, Adeberg S. Treatment Outcome of 227 Patients with Sinonasal Adenoid
Cystic Carcinoma (ACC) after Intensity Modulated Radiotherapy
and Active Raster-Scanning Carbon Ion Boost: A 10-Year Single-Center
Experience. Cancers (Basel) 2019; 111(11): 1705–417.
doi:10.3390/cancers11111705. PMID: 31683896; PMCID:
PMC6895865
- 271 Spiro RH, Huvos AG, Strong EW. Adenoid cystic carcinoma of salivary origin. A clinicopathologic study of 242 cases. Am J Surg 1974; 128: 512-520
- 272 Spiro RH. Salivary neoplasms: Overview of a 35-year experience with 2,807 patients. Head Neck Surg 1986; 8: 177-184
- 273 Michel G, Joubert M, Delemazure AS. et al. Adenoid cystic carcinoma of the paranasal sinuses: Retrospective series and review of the literature. Eur Ann Otorhinolaryngol Head Neck Dis 2013; 130: 257-262
- 274 Kim GE, Park HC, Keum KC. et al. Adenoid cystic carcinoma of the maxillary antrum. Am J Otolaryngol – Head Neck Med Surg 1999; 20: 77-84
- 275 Chen AM, Bucci MK, Weinberg V. et al. Adenoid cystic carcinoma of the head and neck treated by surgery with or without postoperative radiation therapy: Prognostic features of recurrence. Int J Radiat Oncol Biol Phys 2006; 66: 152-159
- 276 Terhaard CHJ, Lubsen H, Rasch CRN. et al. The role of radiotherapy in the treatment of malignant salivary gland tumors. Int J Radiat Oncol Biol Phys 2005; 61: 103-111
- 277 Garden AS, Weber RS, Ang KK. et al. Postoperative radiation therapy for malignant tumors of minor salivary glands. Outcome and patterns of failure. Cancer 1994; 73: 2563-2569
- 278 Chen AM, Granchi PJ, Garcia J. et al. Local-regional recurrence after surgery without postoperative irradiation for carcinomas of the major salivary glands: Implications for adjuvant therapy. Int J Radiat Oncol Biol Phys 2007; 67: 982-987
- 279 Mendenhall WM, Morris CG, Amdur RJ. et al. Radiotherapy alone or combined with surgery for adenoid cystic carcinoma of the head and neck. Head Neck 2004; 26: 154-162
- 280 Terhaard CHJ, Lubsen H, Van Der Tweel I. et al. Salivary gland carcinoma: Independent prognostic factors for locoregional control, distant metastases, and overall survival: Results of the Dutch Head and Neck Oncology Cooperative Group. Head Neck 2004; 26: 681-693
- 281 Katz TS, Mendenhall WM, Morris CG. et al. Malignant tumors of the nasal cavity and paranasal sinuses. Head Neck 2002; 24: 821-829
- 282 Laramore GE, Krall JM, Griffin TW. et al. Neutron versus photon irradiation for unresectable salivary gland tumors: Final report of an RTOG-MRC randomized clinical trial. Int J Radiat Oncol Biol Phys 1993; 27: 235-240
- 283 Douglas JG, Koh WJ, Austin-Seymour M. et al. Treatment of salivary gland neoplasms with fast neutron radiotherapy. Arch Otolaryngol - Head Neck Surg 2003; 129: 944-948
- 284 Stannard C, Vernimmen F, Carrara H. et al. Malignant salivary gland tumours: Can fast neutron therapy results point the way to carbon ion therapy?. Radiother Oncol 2013; 109: 262-268
- 285 Huber PE, Debus J, Latz D. et al. Radiotherapy for advanced adenoid cystic carcinoma: Neutrons, photons or mixed beam?. Radiother Oncol 2001; 59: 161-167
- 286 Pommier P, Liebsch NJ, Deschler DG. et al. Proton beam radiation therapy for skull base adenoid cystic carcinoma. Arch Otolaryngol - Head Neck Surg 2006; 132: 1242-1249
- 287 Patel NR, Sanghvi S, Khan MN. et al. Demografic trends and disease-specific survival in salivary acinic cell carcinoma: An analysis of 1129 cases. Laryngoscope 2014; 124: 172-178
- 288 Haerle SK, Gullane PJ, Witterick IJ. et al. Sinonasal Carcinomas. Epidemiology, Pathology, and Management. Neurosurg Clin N Am 2013; 24: 39-49
- 289 Neto AG, Pineda-Daboin K, Spencer ML. et al. Sinonasal acinic cell carcinoma: A clinicopathologic study of four cases. Head Neck 2005; 27: 603-607
- 290 Wong A, Leong JL, Ho B. Primary acinic cell carcinoma of the ethmoid sinus. Ear, Nose Throat J 2010; 89
- 291 Biron VL, Lentsch EJ, Gerry DR. et al. Case-control analysis of survival outcomes in sinonasal acinic cell carcinoma. Int Forum Allergy Rhinol 2014; 4: 507-511
- 292 Ellis MA, Graboyes EM, Day TA. et al. Prognostic factors and occult nodal disease in mucoepidermoid carcinoma of the oral cavity and oropharynx: An analysis of the National Cancer Database. Oral Oncol 2017; 72: 174-178
- 293 Auger SR, Patel T, Ganti A. et al. Effect of margin status and pathological grade in treatment of sinonasal mucoepidermoid carcinoma. Laryngoscope. 2020
- 294 Bhattacharyya N. Survival and staging characteristics for non-squamous cell malignancies of the maxillary sinus. Arch Otolaryngol - Head Neck Surg 2003; 129: 334-337
- 295 Wolfish EB, Nelson BL, Thompson LDR. Sinonasal Tract Mucoepidermoid Carcinoma: A Clinicopathologic and Immunophenotypic Study of 19 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2012; 6: 191-207
- 296 Lee YS, Ha SM, Paik SW. et al. Epithelial-myoepithelial carcinoma originating from a minor salivary gland in the nasal septum: A case report and literature review. Medicine (Baltimore) 2020; 99: e19072
- 297 Gore MR. Epithelial-myoepithelial carcinoma: A population-based survival analysis. BMC Ear, Nose Throat Disord 2018; 18
- 298 Morresi-Hauf AT, Reu S, Fertl A. Epithelial-myoepitheliales Karzinom der Trachea: Fallbericht und Literaturübersicht. Pathologe 2013; 34: 56-64
- 299 Roh JL, Chang HR, Choi SH. et al. Clinical utility of 18F-FDG PET for patients with salivary gland malignancies. J Nucl Med 2007; 48: 240-246
- 300 Kim CH, Jeong JS, Kim SR. et al. Endobronchial epithelial-myoepithelial carcinoma of the lung. Thorax 2018; 73: 593-594
- 301 Kim SH, Park SE, Bae HG. et al. Epithelial-myoepithelial carcinoma of the nasopharynx: A case report and review of the literature. Oncol Lett 2015; 10: 927-930
- 302 Fried D, Zanation AM, Huang B. et al. Management of nonesthesioneuroblastoma sinonasal malignancies with neuroendocrine differentiation. Laryngoscope 2012; 122: 2210-2215
- 303 Hong SL, Kim SD, Roh HJ. et al. The sphenoid sinus: An unusual presentation of a typical carcinoid tumor. J Craniofac Surg 2014; 25: e483-e485
- 304 Konukiewitz B, Agaimy A, Weichert W. et al. Neuroendokrine Neoplasien der Kopf-Hals-Region. Pathologe 2018; 39: 27-34
- 305 Ferlito A, Silver CE, Bradford CR. et al. Neuroendocrine neoplasms of the larynx: An overview. Head Neck 2009; 31: 1634-1646
- 306 Lee DH, Cho HH, Cho YB. Typical carcinoid tumor of the nasal cavity. Auris Nasus Larynx 2007; 34: 537-539
- 307 Patel TD, Carniol ET, Vázquez A. et al. Sinonasal fibrosarcoma: Analysis of the Surveillance, Epidemiology, and End Results database. Int Forum Allergy Rhinol 2016; 6: 201-205
- 308 Lartigau E, Lusinchi A, Schwaab G. Sarcomas of nasal cavity and paranasal sinuses: Chondrosarcoma, osteosarcoma and fibrosarcoma. J Laryngol Otol 1994; 108: 947-953
- 309 Koshy M, Rich SE, Mohiuddin MM. Improved Survival With Radiation Therapy in High-Grade Soft Tissue Sarcomas of the Extremities: A SEER Analysis. Int J Radiat Oncol Biol Phys 2010; 77: 203-209
- 310 Wang CP, Chang YL, Ting LL. et al. Malignant fibrous histiocytoma of the sinonasal tract. Head Neck 2009; 31: 85-93
- 311 Szablewski V, Neuville A, Terrier P. et al. Adult sinonasal soft tissue sarcoma: Analysis of 48 cases from the French Sarcoma Group database. Laryngoscope 2015; 125: 615-623
- 312 Gerrand CH, Bell RS, Wunder JS. et al. The influence of anatomic location on outcome in patients with soft tissue sarcoma of the extremity. Cancer 2003; 97: 485-492
- 313 Kwok MMK, Lee S, Hosking P. Leiomyosarcoma: A rare sinonasal malignancy. BMJ Case Rep 2018; 2018
- 314 Tanaka H, Westesson PL, Wilbur DC. Leiomyosarcoma of the maxillary sinus: CT and MRI findings. Br J Radiol 1998; 71: 221-224
- 315 Dobben GD. Leiomyosarcoma of the Nasopharynx. AMA Arch Otolaryngol 1958; 68: 211-213
- 316 Sanghvi S, Misra P, Patel NR. et al. Incidence trends and long-term survival analysis of sinonasal rhabdomyosarcoma. Am J Otolaryngol - Head Neck Med Surg 2013; 34: 682-689
- 317 Wurm J, Constantinidis J, Grabenbauer GG. et al. Rhabdomyosarcomas of the nose and paranasal sinuses: Treatment results in 15 cases. Otolaryngol - Head Neck Surg 2005; 133: 42-50
- 318 Narula AA, Vallis MP, El-Silimy OE. et al. Radiation induced angiosarcomas of the nasopharynx. Eur J Surg Oncol 1986; 12: 147-152
- 319 Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: A study of forty-four cases. Cancer 1981; 48: 1907-1921
- 320 Williamson IG, Ramsden RT. Angiosarcoma of maxillary antrum—association with vinyL chloride exposure. J Laryngol Otol 1988; 102: 464-467
- 321 Nelson BL, Thompson LDR. Sinonasal tract angiosarcoma: A clinicopathologic and immunophenotypic study of 10 cases with a review of the literature. Head Neck Pathol 2007; 1: 1-12
- 322 Fukushima K, Dejima K, Koike S. et al. A case of angiosarcoma of the nasal cavity successfully treated with recombinant interleukin-2. Otolaryngol - Head Neck Surg 2006; 134: 886-887
- 323 Treviño-González JL, Santos-Lartigue R, González-Andrade B. et al. Angiosarcoma of the nasal cavity: A case report. Cases J 2009; 2
- 324 Es-Sbissi F, Nitassi S, Boulaadas M. et al. Sinonasal angiosarcoma. Eur Ann Otorhinolaryngol Head Neck Dis 2015; 132: 161-163
- 325 Agaimy A, Hartmann A. Kopf-Hals-Tumoren: Neues aus der WHO-Klassifikation 2017. Pathologe 2018; 39
- 326 Agaimy A, Haller F, Hartmann A. Sinunasale Tumoren: Neues aus der WHO mit besonderem Fokus auf neue mesenchymale Entitäten. Pathologe 2018; 39: 18-26
- 327 Lewis JT, Oliveira AM, Nascimento AG. et al. Low-grade sinonasal sarcoma with neural and myogenic features: A clinicopathologic analysis of 28 cases. Am J Surg Pathol 2012; 36: 517-525
- 328 Rooper LM, Huang SC, Antonescu CR. et al. Biphenotypic sinonasal sarcoma: An expanded immunoprofile including consistent nuclear β-catenin positivity and absence of SOX10 expression. Hum Pathol 2016; 55: 44-50
- 329 Carter CS, East EG, McHugh JB. Biphenotypic sinonasal sarcoma: A review and update. In: Archives of Pathology and Laboratory Medicine. College of American Pathologists 2018; 1196-1201
- 330 Fritchie KJ, Jin L, Wang X. et al. Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases. Histopathology 2016; 69: 930-936
- 331 Ducatman BS, Scheithauer BW, Piepgras DG. et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57: 2006-2021
- 332 Rodriguez FJ, Folpe AL, Giannini C. et al. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012; 123: 295-319
- 333 James AW, Shurell E, Singh A. et al. Malignant Peripheral Nerve Sheath Tumor. Surg Oncol Clin N Am 2016; 25: 789-802
- 334 Bradtmöller M, Hartmann C, Zietsch J. et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS One 2012; 7: e47595
- 335 Kar M, Suryanarayana Deo SV, Shukla NK. et al. Malignant peripheral nerve sheath tumors (MPNST) - Clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol 2006; 4: 55
- 336 Jo VY, Fletcher CDM. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014; 46: 95-104
- 337 Van De Rijn M, Barr FG, Xiong QBin. et al. Radiation-associated synovial sarcoma. Hum Pathol 1997; 28: 1325-1328
- 338 Deraedt K, Debiec-Rychter M, Sciot R. Radiation-associated synovial sarcoma of the lung following radiotherapy for pulmonary metastasis of Wilms’ tumour [7]. Histopathology 2006; 48: 473-475
- 339 Egger JF, Coindre JM, Benhattar J. et al. Radiation-associated synovial sarcoma: Clinicopathologic and molecular analysis of two cases. Mod Pathol 2002; 15: 998-1004
- 340 Sherman KL, Wayne JD, Chung J. et al. Assessment of multimodality therapy use for extremity sarcoma in the United States. J Surg Oncol 2014; 109: 395-404
- 341 Vlenterie M, Jones RL, Van Der Graaf WTA. Synovial sarcoma diagnosis and management in the era of targeted therapies. Curr Opin Oncol 2015; 27: 316-322
- 342 De Bree E, Zoras O, Hunt JL. et al. Desmoid tumors of the head and neck: A therapeutic challenge. Head Neck 2014; 36: 1517-1526
- 343 Orphanet: Desmoid type fibromatosis. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=8665&Disease_Disease_Search_diseaseType=ORPHA&Disease_Disease_Search_diseaseGroup=873&Disease(s)/groupofdiseases=Desmoid-type-fibromatosis&title=Desmoid-type-fibromatosis&search=Dise
- 344 Colombo C, Foo WC, Whiting D. et al. FAP-related desmoid tumors: A series of 44 patients evaluated in a cancer referral center. Histol Histopathol 2012; 27: 641-649
- 345 Coffin CM, Hornick JL, Zhou H. et al. Gardner fibroma: A clinicopathologic and immunohistochemical analysis of 45 patients with 57 fibromas. Am J Surg Pathol 2007; 31: 410-416
- 346 Emori M, Kaya M, Mitsuhashi T. et al. Desmoid tumor-associated pain is dependent on mast cell expression of cyclooxygenase-2. Diagn Pathol 2014; 9
- 347 Kasper B, Baumgarten C, Garcia J. et al. An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol 2017; 28: 2399-2408
- 348 Bonvalot S, Eldweny H, Haddad V. et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 2008; 34: 462-468
- 349 Penel N, Chibon F, Salas S. Adult desmoid tumors: Biology, management and ongoing trials. Curr Opin Oncol 2017; 29: 268-274
- 350 Huang PW, Tzen CY. Prognostic factors in desmoid-type fibromatosis: A clinicopathological and immunohistochemical analysis of 46 cases. Pathology 2010; 42: 147-150
- 351 Granter SR, Badizadegan K, Fletcher CDM. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: A spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol 1998; 22: 513-525
- 352 Asimakopoulos P, Syed MI, Andrews T. et al. Sinonasal glomangiopericytoma: Is anything new?. Ear, Nose Throat J 2016; 95: E1
- 353 Gengler C, Guillou L. Solitary fibrous tumour and haemangiopericytoma: Evolution of a concept. Histopathology 2006; 48: 63-74
- 354 Thompson LDR, Miettinen M, Wenig BM. Sinonasal-type hemangiopericytoma: A clinicopathologic and immunophenotypic analysis of 104 cases showing perivascular myoid differentiation. Am J Surg Pathol 2003; 27: 737-749
- 355 Catalano PJ, Brandwein M, Shah DK. et al. Sinonasal hemangiopericytomas: A clinicopathologic and immunohistochemical study of seven cases. Head Neck 1996; 18: 42-53
- 356 Billings KR, Fu YS, Calcaterra TC. et al. Hemangiopericytoma of the head and neck. Am J Otolaryngol - Head Neck Med Surg 2000; 21: 238-243
- 357 Carew JF, Singh B, Kraus DH. Hemangiopericytoma of the head and neck. Laryngoscope 1999; 109: 1409-1411
- 358 Weber W, Henkes H, Metz KA. et al. Haemangiopericytoma of the nasal cavity. Neuroradiology 2001; 43: 183-186
- 359 Witkin GB, Rosai J. Solitary fibrous tumor of the upper respiratory tract: A report of six cases. Am J Surg Pathol 1991; 15: 842-848
- 360 Batsakis JG, El-Naggar AK, Hybels RD. Pathology consultation solitary fibrous tumor. Ann Otol Rhinol Laryngol 1993; 102: 74-76
- 361 Künzel J, Hainz M, Ziebart T. et al. Head and neck solitary fibrous tumors: a rare and challenging entity. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1589-1598
- 362 Demicco EG, Park MS, Araujo DM. et al. Solitary fibrous tumor: A clinicopathological study of 110 cases and proposed risk assessment model. Mod Pathol 2012; 25: 1298-1306
- 363 Xue Y, Chai G, Xiao F. et al. Post-operative radiotherapy for the treatment of malignant solitary fibrous tumor of the nasal and paranasal area. Jpn J Clin Oncol 2014; 44: 926-931
- 364 Piccaluga PP, De Falco G, Kustagi M. et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood 2011; 117: 3596-3608
- 365 Chi AC, Weathers DR, Folpe AL. et al. Epithelioid hemangioendothelioma of the oral cavity: Report of two cases and review of the literature. Oral Surgery, Oral Med Oral Pathol Oral Radiol Endodontology 2005; 100: 717-724
- 366 Errani C, Zhang L, Sung YS. et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosom Cancer 2011; 50: 644-653
- 367 Flucke U, Vogels RJC, de Saint Aubain Somerhausen N. et al. Epithelioid Hemangioendothelioma: Clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol 2014; 9
- 368 Bruder E, Alaggio R, Kozakewich HPW. et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol 2012; 15: 26-61
- 369 Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma a vascular tumor often mistaken for a carcinoma. Cancer 1982; 50: 970-981
- 370 Mentzel T, Beham A, Calonje E. et al. Epithelioid hemangioendothelioma of skin and soft tissues: Clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 1997; 21: 363-374
- 371 Deyrup AT, Tighiouart M, Montag AG. et al. Epithelioid hemangioendothelioma of soft tissue: A proposal for risk stratification based on 49 cases. Am J Surg Pathol 2008; 32: 924-927
- 372 Beger M, Antoni C, Haas S. et al. Epitheloides Hämangioendotheliom- drei Patienten, drei Therapieoptionen. Z Gastroenterol 2006; 44: A5_04
- 373 Wong BLK, Lee VNY, Tikka T. et al. Kaposiform haemangioendothelioma of the head and neck. Crit Rev Oncol Hematol 2016; 104: 156-168
- 374 Sun ZJ, Zhang L, Zhang WF. et al. Kaposiform hemangioendothelioma involving the neck. Oral Oncol Extra 2006; 42: 60-65
- 375 Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis 2013; 8: 95
- 376 Pongpruttipan T, Sukpanichnant S, Assanasen T. et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and αβ, γδ, and αβ/γδ T-cell origin: A comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol 2012; 36: 481-499
- 377 Jhuang JY, Chang ST, Weng SF. et al. Extranodal natural killer/T-cell lymphoma, nasal type in Taiwan: A relatively higher frequency of T-cell lineage and poor survival for extranasal tumors. Hum Pathol 2015; 46: 313-321
- 378 De Campos-Lima PO, Gavioli R, Zhang QJ. et al. HLA-A11 epitope loss isolates of Epstein-Barr virus from a highly A11 + population. Science (80-) 1993; 260: 98-100
- 379 Lima PODC, Levitsky V, Brooks J. et al. T cell responses and virus evolution: Loss of hla all-restricted ctl epitopes in epstein-barr virus isolates from highly all-positive populations by selective mutation of anchor residues. J Exp Med 1994; 179: 1297-1305
- 381 Kim SJ, Choi JY, Hyun SH. et al. Risk stratification on the basis of Deauville score on PET-CT and the presence of Epstein-Barr virus DNA after completion of primary treatment for extranodal natural killer/T-cell lymphoma, nasal type: A multicentre, retrospective analysis. Lancet Haematol 2015; 2: e66-e74
- 381 Tse E, Kwong YL. How i treat NK/T-cell lymphomas. Blood 2013; 121: 4997-5005
- 382 Kwong YL, Kim WS, Lim ST. et al. SMILE for natural killer/T-cell lymphoma: Analysis of safety and efficacy from the Asia Lymphoma Study Group. Blood 2012; 120: 2973-2980
- 383 Alexiou C, Kau RJ, Dietzfelbinger H. et al. Extramedullary plasmacytoma: Tumor occurrence and therapeutic concepts. Cancer 1999; 85: 2305-2314
- 384 Miller FR, Lavertu P, Wanamaker JR. et al. Plasmacytomas of the head and neck. Otolaryngol - Head Neck Surg 1998; 119: 614-618
- 385 Patel TD, Vázquez A, Choudhary MM. et al. Sinonasal extramedullary plasmacytoma: A population-based incidence and survival analysis. Int Forum Allergy Rhinol 2015; 5: 862-869
- 386 Soutar R, Lucraft H, Jackson G. et al. Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. Br J Haematol 2004; 124: 717-726
- 387 Lorsbach RB, Hsi ED, Dogan A. et al. Plasma cell myeloma and related neoplasms. In: American Journal of Clinical Pathology 2011; 168-182
- 388 Susnerwala SS, Shanks JH, Banerjee SS. et al. Extramedullary plasmacytoma of the head and neck region: Clinicopathological correlation in 25 cases. Br J Cancer 1997; 75: 921-927
- 389 Hussong JW, Perkins SL, Schnitzer B. et al. Extramedullary plasmacytoma: A form of marginal zone cell lymphoma?. Am J Clin Pathol 1999; 111: 111-116
- 390 Boll M, Parkins E, O’Connor SJM. et al. Extramedullary plasmacytoma are characterized by a „myeloma-like“ immunophenotype and genotype and occult bone marrow involvement. Br J Haematol 2010; 151: 525-527
- 391 Galieni P, Cavo M, Pulsoni A. et al. Clinical outcome of extramedullary plasmacytoma. Haematologica 2000; 85: 47-51
- 392 Sasaki R, Yasuda K, Abe E. et al. Multi-institutional analysis of solitary extramedullary plasmacytoma of the head and neck treated with curative radiotherapy. Int J Radiat Oncol Biol Phys 2012; 82: 626-634
- 393 Knowling MA, Harwood AR, Bergsagel DE. Comparison of extramedullary plasmacytomas with solitary and multiple plasma cell tumors of bone. J Clin Oncol 1983; 1: 255-262
- 394 Hamre M, Hedberg J, Buckley J. et al. Langerhans cell histiocytosis: An exploratory epidemiologic study of 177 cases. Med Pediatr Oncol 1997; 28: 92-97
- 395 Laman JD, Leenen PJM, Annels NE. et al. Langerhans-cell histiocytosis „insight into DC biology“. Trends Immunol 2003; 24: 190-196
- 396 Orphanet: Langerhans Zell Histiozytose. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=389
- 397 Kostyra K, Kostkiewicz B. Langerhans cell histiocytosis of the orbit and frontal sinus of the adult woman: A first case report in Poland. Surg Neurol Int 2019; 10: 234
- 398 Mani S, Thomas R, Mathew J. et al. Langerhan’s cell histiocytosis of sphenoid sinus causing vision loss: A case report. J Nepal Med Assoc 2019; 57: 281-284
- 399 Gündüz K, Palamar M, Parmak N. et al. Eosinophilic granuloma of the orbit: Report of two cases. J AAPOS 2007; 11: 506-508
- 400 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthal Plast Reconstr Surg 2016; 32: 28-34
- 401 Prayer D, Grois N, Prosch H. et al. MR imaging presentation of intracranial disease associated with langerhans cell histiocytosis. Am J Neuroradiol 2004; 25: 880-891
- 402 Allen CE, Li L, Peters TL. et al. Cell-Specific Gene Expression in Langerhans Cell Histiocytosis Lesions Reveals a Distinct Profile Compared with Epidermal Langerhans Cells. J Immunol 2010; 184: 4557-4567
- 403 Hyman DM, Diamond EL, Vibat CRT. et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015; 5: 64-71
- 404 Berres ML, Lim KPH, Peters T. et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014; 211: 669-683
- 405 Wladis EJ, Tomaszewski JE, Gausas RE. Langerhans cell histiocytosis of the orbit 10 years after involvement at other sites. Ophthal Plast Reconstr Surg 2008; 24: 142-143
- 406 Herwig MC, Wojno T, Zhang Q. et al. Langerhans Cell Histiocytosis of the Orbit: Five Clinicopathologic Cases and Review of the Literature. Surv Ophthalmol 2013; 58: 330-340
- 407 Windfuhr JP. Primitive neuroectodermal tumor of the head and neck: Incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 2004; 113: 533-543
- 408 Hafezi S, Seethala RR, Stelow EB. et al. Ewing’s Family of Tumors of the Sinonasal Tract and Maxillary Bone. Head Neck Pathol 2011; 5: 8-16
- 409 Specht K, Sung YS, Zhang L. et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged ewing sarcomas: Further evidence toward distinct pathologic entities. Genes Chromosom Cancer 2014; 53: 622-633
- 410 Lin JK, Liang J. Sinonasal Ewing Sarcoma: A Case Report and Literature Review. Perm J 2018; 22
- 411 Allam A, El-Husseiny G, Khafaga Y. et al. Ewing’s sarcoma of the head and neck: A retrospective analysis of 24 cases. Sarcoma 1999; 3: 11-15
- 412 Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010; 11: 184-192
- 413 Broich G, Pagliari A, Ottaviani F. Esthesioneuroblastoma: A general review of the cases published since the discovery of the tumour in 1924. Anticancer Res 1997; 17: 2683-2706
- 414 Roussel LM, Patron V, Maubert E. et al. New landmarks in endonasal surgery: from nasal bone to anterior cribriform plate including branches of anterior ethmoidal artery and nerve and terminal nerve. Int Forum Allergy Rhinol 2020; 10: 395-404
- 415 Mills SE. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 2002; 15: 264-278
- 416 Thompson LDR. Olfactory Neuroblastoma. Head Neck Pathol 2009; 3: 252-259
- 417 Kadish S, Goodman M, Wang CC. Olfactory neuroblastoma—A clinical analysis of 17 cases. Cancer 1976; 37: 1571-1576
- 418 Morita A, Ebersold MJ, Olsen KD. et al. Esthesioneuroblastoma: Prognosis and management. Neurosurgery 1993; 32: 706-715
- 419 Edge SB, Compton CC. The american joint committee on cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010; 17: 1471-1474
- 420 Norton A. Tumors of the Upper Respiratory Tract and Ear. In: Hyams V, Batsakis J, Michaels L Hrsg. Journal of Clinical Pathology 1989; 335-335
- 421 Mao L, Xia YP, Zhou YN. et al. Activation of sonic hedgehog signaling pathway in olfactory neuroblastoma. Oncology 2009; 77: 231-243
- 422 Ketcham AS, Wilkins RH, Van Buren JM. et al. A combined intracranial facial approach to the paranasal sinuses. Am J Surg 1963; 106: 698-703
- 423 Hanna E, DeMonte F, Ibrahim S. et al. Endoscopic resection of sinonasal cancers with and without craniotomy: Oncologic results. Arch Otolaryngol - Head Neck Surg 2009; 135: 1219-1224
- 424 Su SY, Bell D, Ferrarotto R. et al. Outcomes for olfactory neuroblastoma treated with induction chemotherapy. Head Neck 2017; 39: 1671-1679
- 425 Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: A meta-analysis and review. Lancet Oncol 2001; 2: 683-690
- 426 Ow TJ, Hanna EY, Roberts DB. et al. Optimization of long-term outcomes for patients with esthesioneuroblastoma. Head Neck 2014; 36: 524-530
- 427 Almutuawa DM, Strohl MP, Gruss C. et al. Outcomes of sinonasal mucosal melanomas with endoscopic and open resection: a retrospective cohort study. J Neurooncol. 2020
- 428 Thompson LDR, Wieneke JA, Miettinen M. Sinonasal tract and nasopharyngeal melanomas: A clinicopathologic study of 115 cases with a proposed staging system. Am J Surg Pathol 2003; 27: 594-611
- 429 Moreno MA, Roberts DB, Kupferman ME. et al. Mucosal melanoma of the nose and paranasal sinuses, a contemporary experience from the M. D. Anderson cancer center. Cancer 2010; 116: 2215-2223
- 430 Patel SG, Prasad ML, Escrig M. et al. Primary mucosal malignant melanoma of the head and neck. Head Neck 2002; 24: 247-257
- 431 Zebary A, Jangard M, Omholt K. et al. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: A study of 56 cases. Br J Cancer 2013; 109: 559-564
- 432 Van Essen TH, Van Pelt S, Versluis M. et al. Prognostic parameters in uveal melanoma and their association with bap1 expression. Br J Ophthalmol 2014; 98: 1738-1743
- 433 Dauer EH, Lewis JE, Rohlinger AL. et al. Sinonasal melanoma: A clinicopathologic review of 61 cases. Otolaryngol - Head Neck Surg 2008; 138: 347-352
- 434 Bachar G, Kwok SL, O’Sullivan B. et al. Mucosal melanomas of the head and neck: The Princess Margaret Hospital experience. Head Neck 2008; 30: 1325-1331
- 435 Lund VJ, Chisholm EJ, Howard DJ. et al. Sinonasal malignant melanoma: An analysis of 115 cases assessing outcomes of surgery, postoperative radiotherapy and endoscopic resection. Rhinology 2012; 50: 203-210
- 436 Thierauf J, Glück AM, Plinkert P. et al. Mucosal melanoma of the cranio-facial region: Surgical challenges and therapeutic options. Auris Nasus Larynx 2019; 46: 252-259
- 437 Gore MR, Zanation AM. Survival in sinonasal melanoma: A meta-analysis. J Neurol Surgery, Part B Skull Base 2012; 73: 157-162
- 438 Ganly I, Patel SG, Singh B. et al. Craniofacial resection for malignant melanoma of the skull base: Report of an International Collaborative Study. Arch Otolaryngol - Head Neck Surg 2006; 132: 73-78
- 439 Jarrom D, Paleri V, Kerawala C. et al. Mucosal melanoma of the upper airways tract mucosal melanoma: A systematic review with meta-analyses of treatment. Head Neck 2017; 39: 819-825
- 440 Li J, Kan H, Zhao L. et al. Immune checkpoint inhibitors in advanced or metastatic mucosal melanoma: a systematic review. Ther Adv Med Oncol 2020; 12
- 441 Misra P, Husain Q, Svider PF. et al. Management of sinonasal teratocarcinosarcoma: A systematic review. Am J Otolaryngol - Head Neck Med Surg 2014; 35: 5-11
- 442 Chao KK, Eng TY, Barnes J. et al. Sinonasal Teratocarcinosarcoma. Am J Clin Oncol Cancer Clin Trials 2004; 27: 29-32
- 443 Fukuoka K, Hirokawa M, Shimizu M. et al. Teratocarcinosarcoma of the nasal cavity. Report of a case showing favorable prognosis. Apmis 2000; 108: 553-557
- 444 Heffner DK, Hyams VJ. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses: A clinicopathologic study of 20 cases. Cancer 1984; 53: 2140-2154
- 445 Wei S, Carroll W, Lazenby A. et al. Sinonasal teratocarcinosarcoma: report of a case with review of literature and treatment outcome. Ann Diagn Pathol 2008; 12: 415-425
- 446 Vranic S, Caughron SK, Djuricic S. et al. Hamartomas, teratomas and teratocarcinosarcomas of the head and neck: Report of 3 new cases with clinico-pathologic correlation, cytogenetic analysis, and review of the literature. BMC Ear, Nose Throat Disord 2008; 8
- 447 Lim CCT, Thiagarajan A, Sim CS. et al. Craniospinal dissemination in teratocarcinosarcoma: Case report. J Neurosurg 2008; 109: 321-324
- 448 Terasaka S, Medary MB, Whiting DM. et al. Prolonged survival in a patient with sinonasal teratocarcinosarcoma with cranial extension. Case report. J Neurosurg 1998; 88: 753-756
- 449 Dicke TE, Gates GA. Malignant Teratoma of the Paranasal Sinuses: Report of a Case. Arch Otolaryngol 1970; 91: 391-394