

Subscribe to RSS
DOI: 10.1055/a-2722-5684
Hepatocellular Carcinoma – from Immunobiology to Immunotherapy
HCC – von der Immunbiologie zur ImmuntherapieAuthors
Supported by: Deutsches Konsortium für Translationale Krebsforschung
Supported by: Deutsche Forschungsgemeinschaft 272983813 ,390939984 ,441891347,520992132

Abstract
Hepatocellular Carcinoma (HCC) is an entity characterized by a highly heterogenous tumor immune microenvironment. The introduction of immune checkpoint inhibitor (ICI) therapy as standard of care in advanced disease stages and with promising results in earlier stages has highlighted the need for a better understanding of the underlying immunobiology. In this review, we provide a summary about the immune landscape in HCC and discuss novel potential therapeutic targets as well as how spatial immune profiling may help identify optimal candidates for ICI therapy.
Zusammenfassung
Das Hepatozelluläre Karzinom (HCC) zeichnet sich durch ein heterogenes Tumorimmunmilieu aus. Die Einführung von Immuncheckpointinhibitoren (ICI) als Standardtherapie bei fortgeschrittenem HCC sowie als vielversprechende Option in früheren Krankheitsstadien hebt die Bedeutung eines besseren Verständnisses der zugrunde liegenden Tumorimmunologie hervor. Diese Übersichtsarbeit fasst die immunologische Landschaft beim HCC zusammen, stellt potenzielle neue Therapieoptionen dar und geht im Besonderen auf die Bedeutung von räumlichen Analysen bei der Auswahl geeigneter Patienten für Immuntherapien ein.
Schlüsselwörter
Leber - Onkologie - Immuntherapie - Immunologie - Hepatozelluläres Karzinom - ImmunotypIntroduction
Hepatocellular carcinoma (HCC) represents a major global health challenge ranking as the third most common cause of cancer-related death with an incidence in Germany estimated to be around 9800 cases/year [1]. HCC usually develops in patients with chronic liver disease, such as chronic viral hepatitis and liver cirrhosis. Treatment decisions for HCC are traditionally guided by the Barcelona Clinic for liver cancer (BCLC) classification based on tumor size, number of nodules, spread and liver function [2] [3] ([Fig. 1]). Resection, liver transplantation or local ablation are preferred choices in the early, curative stages. Locoregional therapies such as transarterial chemoembolization (TACE) and transarterial radioembolization (TARE) or stereotactic radiotherapy represent the backbone in intermediate stage BCLC B. Systemic therapy is recommended in the advanced stage BCLC C if the cancer shows extrahepatic spread or macrovascular invasion. In these patients, systemic immune checkpoint inhibitor (ICI)-based therapies have shown improved efficacy and survival over tyrosine kinase Inhibitor (TKI) therapies that were previously used when other therapeutic options were exploited. However, in the light of expanding options of ablative and locoregional therapies and an efficacy of systemic immune checkpoint inhibitor (ICI)-based therapies also in earlier disease stages, the traditional BCLC stage-guided treatment recommendations require careful reconsideration. For example, treatment stage migration may be indicated upon progression after the first treatment choice [3]. Moreover, the application of immune-based therapy approaches may benefit from a careful evaluation of the underlying immunobiology. This review will discuss the current understanding of the immunobiology of HCC and how it may influence the decision making for immunotherapy regimen.

History and current status of HCC systemic therapy
The oral multikinase inhibitor sorafenib represented the mainstay of systemic therapy of advanced HCC for more than a decade. In 2007, in the phase III randomized controlled SHARP trial sorafenib demonstrated a 2.8 month median overall survival benefit versus placebo [4]. This therapeutic effect was confirmed in an Asian patient cohort [5]. However, the therapeutic use remained limited, as a) only patients with preserved liver function and a sufficient ECOG status were suitable for sorafenib; b) no combination treatment approaches with surgery or locoregional therapies became established and c) sorafenib caused severe tyrosinkinase inhibitor (TKI)-typical side effects such as diarrhea, weight loss and hand-foot reaction. Nevertheless, sorafenib was the sole approved therapy for BCLC stage C until 2018, when another broad multikinase inhibitor, lenvatinib, showed non-inferiority to sorafenib in the REFLECT trial [6]. Lenvatinib even displayed slightly superior results in the endpoints objective response rates, progression-free survival and time to progression. This was followed by approvals of additional second line therapies, such as the TKIs regorafenib and cabozantinib and the anti-VEGFR2 antibody ramucirumab.
Following the success of ICI in other cancers, checkpoint inhibitor regimen targeting programmed cell death protein 1 (PD-1), its ligand PD-L1, and cytotoxic T-lymphocyte antigen 4 (CTLA-4) were tested in HCC. Despite early indications of efficacy of anti-PD-1 based monotherapies, somewhat optimistically designed phase III trials did not meet their final statistical endpoints. However, the combination therapy regimen combining anti-PD-L1 with anti-VEGF therapy or using different ICIs combinations has demonstrated superiority over TKI based therapies. Thus, since 2020, combination ICI therapies represent the first-line therapy for HCC. In the following we provide a brief summary of the data for approved and promising therapies in HCC.
Nivolumab is a monoclonal antibody that blocks the inhibitory binding of PD-L1 to PD-1 receptors expressed on T cells. It is widely used in multiple malignancies and first demonstrated efficacy in malignant melanoma and non-small cell lung cancer (NSCLC). Based on the phase II study CheckMate 040, which showed an overall survival rate of 72% at 6 months and a response rate of 20% in patients after prior sorafenib therapy, Nivolumab received an accelerated FDA-approval as second line treatment in 2017 [7]. However, the phase 3 trial CheckMate-459 only demonstrated a non-significant trend for a survival benefit compared to sorafenib (OS HR 0.84, p = 0.04) [8]. Nivolumab monotherapy exhibited a spectrum of side effects typical for ICI therapies, including immune-mediated hepatitis, colitis or pneumonitis. Similar observations were made in clinical trials testing the anti-PD1-antibody pembrolizumab in second line after sorafenib that after promising phase II trial data with a 17% response rate [9] could not meet its prespecified primary endpoints in the phase 3 trial Keynote-240 [10].
Finally, the approval of the PD-L1 inhibitor atezolizumab represented a small revolution in the systemic treatment of HCC. Atezolizumab was tested in the IMBRAVE-150 study in combination with the VEGF inhibitor bevacizumab and showed an improved overall survival (67.2% vs. 54.6% at 12 months) and progression-free survival (6.8 months vs. 4.3 months) compared to sorafenib [11]. This therapeutic approach combining immunotherapy with anti-angiogenetic therapy became the new standard for the systemic treatment of advanced HCC in BCLC stage C. In addition to the anticipated profile of ICI-related adverse effects, a higher bleeding risk attributed to the anti-VEGF therapy requires careful consideration.
The HIMALAYA trial, published 2022, led to the first approval of a “pure” immunotherapy regimen. The three-arm study with tremelimumab, a blocking anti-CTLA-4 antibody, in combination with durvalumab, an anti-PD-L1 antibody, using the infusion regimen “STRIDE” (Single Tremelimumab Regular Interval Durvalumab) vs. durvalumab monotherapy vs. sorafenib showed overall survival rates at 36 months of 30.7%, 24.7% and 20.2%, respectively [12], demonstrating superiority over sorafenib. The STRIDE regimen was subsequently approved as an alternative first-line therapy – and became a preferred choice for patients with relative contraindications to bevacizumab therapy, such as wound healing disorders or high bleeding risk. Nevertheless, dual ICI therapy is also associated with significant rates of immune-mediated side effects. Durvalumab monotherapy also received approval in first-line therapy of patients with advanced HCC and is recommended in patients that are not eligible for combination regimens.
In March 2025, based on results from the CheckMate-9DW study, the combined immunotherapy regimen of nivolumab and ipilimumab was approved by the EMA. Ipilimumab is a widely used anti-CTLA-4 antibody [13] and patients receive up to four doses of anti-CTLA-4 therapy while nivolumab therapy is continued after the fourth cycle. The combination was tested against sorafenib or lenvatinib. The study showed overall response rates of 36% versus 13% and a median overall survival advantage for nivolumab + ipilimumab of 23.7 months vs. 20.6 months. Treatment-related adverse events (TRAEs) were comparable in both groups: any TRAEs 84% for nivolumab + ipilimumab vs. 91% for sorafenib or lenvatinib and grade 3–4 TRAEs in 41% vs. 42%, respectively. An overview about current systemic treatment options for patients with advanced HCC is given in [Fig. 2].

Other substances that have been investigated in phase III studies include tislelizumab, cabozantinib plus atezolizumab, camrelizumab plus rivoceranib and lenvatinib plus pembrolizumab. Tislelizumab, an anti-PD-1 blocking antibody demonstrated non-inferiority, a higher objective response rate (14.3% vs. 5.4%) and a more durable response compared to sorafenib in the RATIONALE-301 trial [14]. The COSMIC-312 trial combined atezolizumab with cabozantinib, a TKI that was already approved for second-line therapy. The results were disappointing, since the results for the primary endpoint median OS were comparable to that of patients treated with sorafenib (15.4 months vs. 15.5 months) [15]. A comparable result was obtained for the combination of lenvatinib with pembrolizumab in the LEAP-002 study: the addition of pembrolizumab to lenvatinib compared to lenvatinib plus placebo did not meet significance for the endpoints overall survival (21.2 vs. 19.0 months) and progression-free survival (8.2 vs. 8.0 months) [16]. However, camrelizumab (anti-PD-1) plus rivoceranib (TKI) vs. sorafenib in the CARES-310 study demonstrated a significant improvement for its primary endpoint progression-free survival (5.6 vs. 3.7 months) as well as for median overall survival (22.1 vs. 15.2 months) [17]. However, combination therapies with TKIs are currently not approved.
Although ICI therapies can lead to complete and long-lasting responses even in advanced stages of disease [18], clinical trials have demonstrated that only a fraction of patients responds to current therapy regimen. Indeed, objective response rates (ORR) in the approved therapy regimen range from 17–36%. This has been associated with a generally immunotolerant microenvironment in the liver favoring homeostasis and reducing inflammation [19]. In healthy livers, this immune environment ensures tolerance towards gut-derived bacterial products and metabolites and nutrients derived from food intake. This predisposition towards immune tolerance may contribute to promoting persistent viral hepatitis or tumor progression [20]. It may also impede the response to ICI therapies of liver cancer. The exact interplay between different intrahepatic cell types and their environment is only partly understood. A thorough understanding of the HCC tumor microenvironment (TME) and identification of biomarkers to guide ICI-based therapies is thus essential for evidence-based treatment decisions. A precise understanding of the TME may further allow interventions to maximize treatment efficacy and minimize side effects of ICI in HCC.
Immunobiology of HCC and the tumor microenvironment
HCC is a highly heterogeneous tumor that can exhibit a plethora of morphologic features or molecular subtypes, and is also heterogeneous with respect to immune infiltration [21]. Particularly immunosuppressive and immunoregulatory cell types, such as regulatory T cells (Tregs) and tumor-associated macrophages are enriched in the TME of HCC and contribute to tumor immune evasion ([Fig. 3]). In contrast, immune effector cells, such as CD8+ T cells can mediate anti-tumor effects and represent the main targets for ICI. Factors influencing the tumor immune microenvironment include the tumor mutational profile [22] or the underlying etiology of liver disease [23]. In the following sections, we discuss the role of innate and adaptive immune cells and stromal components in the HCC TME and discuss the knowns and unknowns of their contributions to ICI therapy responses.
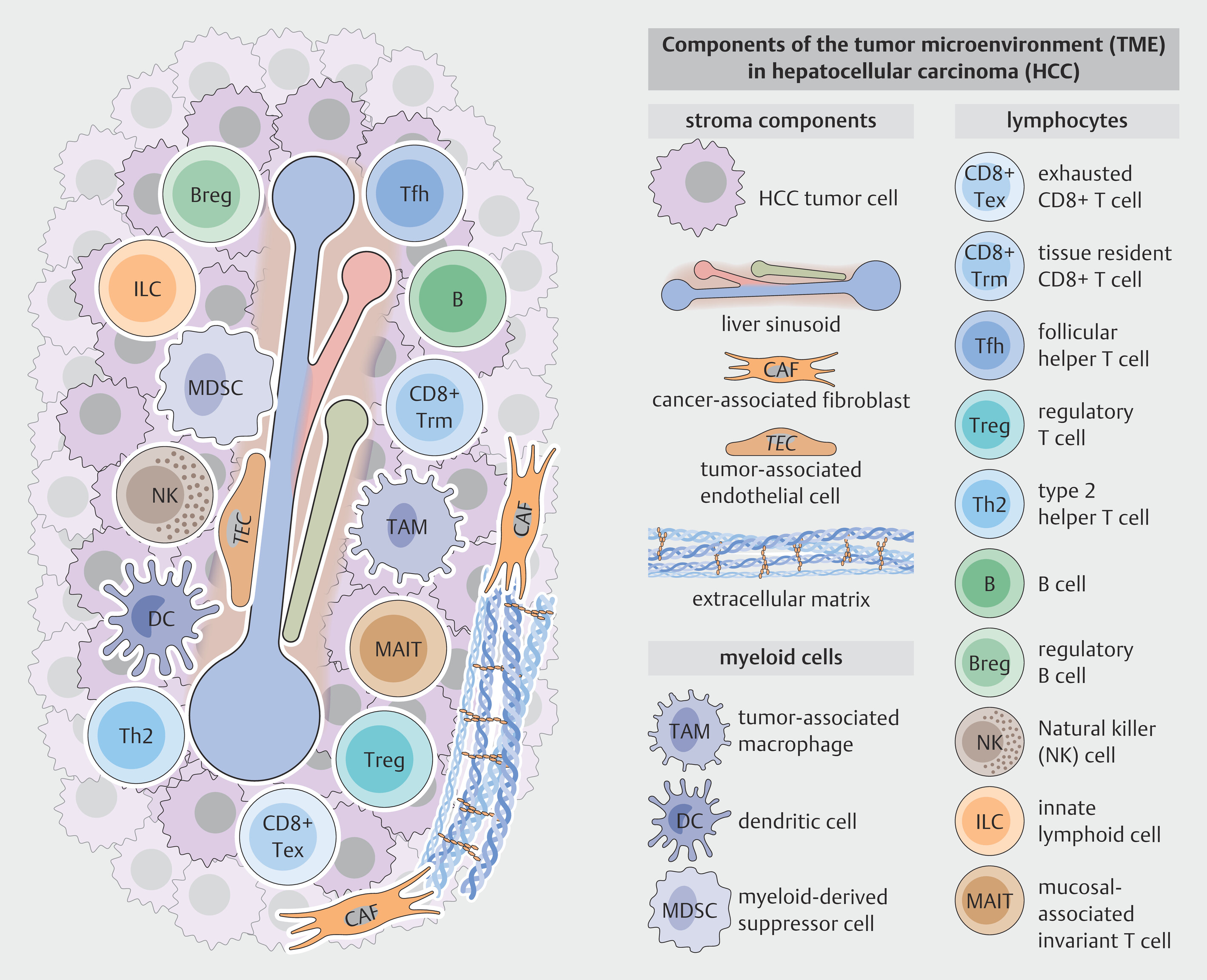
Myeloid cells restrict antitumor immune surveillance
Myeloid cells constitute the most abundant innate immune population within the tumor microenvironment (TME). Well-characterized populations in the TME include tumor-associated macrophages (TAMs), myeloid derived suppressor cells (MDSCs) and dendritic cells (DCs).
Tumor-associated macrophages
TAMs in HCC are a heterogenous cell population and can originate from diverse precursors. A recent single cell RNA sequencing (scRNAseq) study identified 11 subtypes of intratumoral macrophages that constituted approximately 46% of immune cells in HCC. Interestingly, the majority of TAMs appears to be derived from monocytes and are characterized by markers such as TREM2, VEGFA, MMP9, SPP1 and ITGAM. Only a small fraction of TAMs derive from liver resident Kupffer cells [24]. The recruitment of peripheral blood monocytes to the liver is driven by chemokines, such as CCL2, CX3CL1 and VEGF that are abundant in chronic inflammatory liver and that are further strongly secreted by tumor cells and its microenvironment in HCC [25] [26]. Cancer-associated fibroblasts further recruit monocytes via expression of endosialin that interacts with CD68 on monocytes [27]. Transendothelial migration of monocytes is facilitated by endothelial expression of adhesion molecules such as PLVAP and CD31. Differentiation of transmigrated monocytes into macrophages and its polarization towards TAM’s is promoted by cytokines and growth factors, such as TGFβ and IL-10 secreted by HCC tumor cells and stromal cells [28]. Moreover, metabolic influences, such as hypoxia or metabolites such as α-ketoglutarate promote macrophage polarization towards an immunosuppressive TAM phenotype [29] [30]. Interestingly TAMs have further been reported to secrete HGF that supports M2 polarization in an autocrine manner. Overall the majority of TAMs shows an immunosuppressive phenotype with upregulation of M2 markers such as CD163 and CD206, immune checkpoint molecules (PD-L1 and CD73) and an altered metabolism and signaling [31]. However, existence of transitional cell states with expression of both M1 and M2 markers stress the classical M1/M2 polarization theory and underline the remarkable plasticity of macrophages. Among the heterogenous TAM population in HCC, particularly SPP1+, TREM2+ and MMP9+ TAMs have been studied in the context of immune evasion. SPP1+ TAMs are often found at the tumor margin in co-localisation with cancer stem cells (CSCs) and their frequency has been associated with tumor stemness, resistance to immunotherapy and overall poor prognosis. Mechanistically, binding of SPP1 to its receptor CD44 on tumor cells and other immune cells promotes survival of CSCs on the one hand and T cell exhaustion and decreased function of CD8+ T cells on the other [32]. TREM2+ TAMs represent a subpopulation of highly immunosuppressive macrophages, that recruit Tregs and directly suppress infiltration and function of CD8+ cytotoxic T cells [33] [34]. In fact, TREM2+ macrophage infiltration is an independent negative prognostic biomarker in HCC [34]. MMP9+ TAMs have been associated with extracellular matrix remodeling, tumor invasion and angiogenesis and are primarily localized at the tumor capsules [31] [35]. By activation of pro-tumoral signaling pathways, such as the PPARγ pathway these cells further directly support tumor growth and progression. Taken together, TAMs represent one of the most abundant immunosuppressive cell population within the tumor microenvironment of HCC.
MDSCs
Myeloid derived suppressor cells (MDSCs) derive from common myeloid progenitors in the bone marrow and represent immature myeloid cells emerging in the context of chronic inflammatory diseases or tumors [36]. MDSCs are characterized by high expression of immunosuppressive factors, such as arginase 1 or reactive oxygen [37] [38] and resemble either monocytes (M-MDSCs) or neutrophils (PMN-MDSCs) [39]. Importantly, differentiation of MDSCs from their myeloid counterparts is challenging, therefore, reliable identification as MDSC requires validation of its immunosuppressive phenotype [40]. Studies have demonstrated an increased frequency of circulating CD14+HLA-DR− MDSCs in HCC [41]. Moreover, MDSC’s are increasingly recruited to HCC tumor sites and its accumulation in tumor tissue correlates with poor prognosis [42] [43]. Multiple chemokines secreted by the tumor cell itself and its microenvironment, such as CAFs or macrophages are involved in recruitment of MDSCs. For instance, CAFs secrete CCL2 that binds on CCR2 on MDSCs facilitating their migration to tumor sites [44]. Moreover, tumor cell derived CCL26 has been shown to promote recruitment of CX3CR1+ MDSCs to HCC [45]. Functionally, MDSCs promote immunosuppression in HCC. MDSCs strongly express arginase 1 and iNOS involved in depleting the essential amino acid L-Arginine required for T cell function which can induce impaired CD4+ T-cell proliferation and activation. MDSCs can further suppress T cell responses by induction of Tregs [41]. Moreover, MDSCs produce high levels of reactive oxygen species and NO that inhibit T cell function [46] [47] [48]. Finally, MDSCs can inhibit NK cell cytotoxicity via NKp30 mediated direct cell contact [49] and reduce the antigen-presenting function of Kupffer cells by increasing PD-L1 expression and decreasing the levels of CD86 and MHCII on their surface [50]. Taken together, MDSCs may strongly contribute to immune evasion and tumor progression in HCC. The link between MDSC infiltration, poorer clinical outcome and reduced treatment response [51] makes these cells attractive targets for novel therapeutic concepts.
Dendritic cells
Dendritic cells encompass a diverse population of specialized antigen-presenting cells that play a crucial role in initiating T cell activation, hereby linking innate and adaptive immunity [52]. DCs can be divided into three main subclasses: conventional DCs type 1 (cDC1), conventional DCs type 2 (cDC2), plasmacytoid DCs and monocyte derived DCs. Patients with HCC frequently show a decrease of overall DC frequency [53]. Functionally, cDC1 have been described to primarily mediate anti-tumor immunity by cross-presenting tumor antigens to activate CD8+ T cells. In line with this, high cDC1 levels in HCC are linked to better prognosis. cDC2 appear to mediate dual functions in tumor immune control. Thus, cDC2 can promote anti-tumor immune effects by presenting tumor antigens to naive CD4+ T cells and further promoting their differentiation into Th1 or Th17 effector cells by secretion of IL-12 and IL-23 [54]. The immunosuppressive and hypoxic tumor niche in HCC, however, can lead to dysfunctional cDC2s that fail to properly stimulate T cells and rather acquire a TAM-like phenotype, thus contributing to immune tolerance und tumor immune evasion [54] [55]. These dysfunctional cDCs2 show impaired antigen presentation and are associated with the recruitment of Tregs [56]. Plasmacytoid DCs are the main producers of interferons and can activate CD8+ T cells and NK cells. In HCC, these cells are often dysfunctional and show reduced Interferon I production. One study further reported pDCs to foster an immunosuppressive microenvironment in relapsing HCC after surgery by recruiting MDSCs via IFNα and the IRF1/CX3CL1 signaling pathway [57]. In sum, impairment of DC function contributes to immunosuppression in HCC.
Lymphocytes in HCC
B and T lymphocytes represent the major immune cells of the adaptive immune system that can recognize specific (tumor) antigens using specific rearranged antigen receptors (B cell receptor (BCR) and T cell receptor (TCR), respectively). While B cells contribute to humoral immunity as antigen-presenting cells and potential precursors of antibody-producing plasma cells, CD8+ and CD4+ T cells can recognize antigens presented on HLA molecules e.g. via antigen-presenting cells (APCs) and exert direct antitumor functions through cytolytic and non-cytolytic functions. However, in the tumor context, many CD8+ T cells express immune checkpoints such as PD-1, and fail to maintain effector function and are characterized by exhausted and dysfunctional programs. Moreover, CD4+ T cells can polarize into immunoregulatory populations, such as CTLA-4 expressing FoxP3+ regulatory T cells, effectively supporting tumor growth. Other lymphocytes comprise the group of innate or innate-like lymphocytes, with limited recall capacities but important direct antitumor effector functions as potential targets for future therapies.
B cells: conflicting roles in HCC
Alongside with other immune cells, B cells are frequently located at the tumor margin and remain scarce in the tumor center [58]. In human HCC, high infiltration of B cell infiltration has been associated with better survival of patients [58] [59] [60] [61].
Other reports, however, found infiltration of B cells and plasma cells to be associated with less differentiated tumors and poor prognosis [62] [63]. More particularly, IL-10 and TGF-β producing B cells and their potential precursors have been implicated in tumor progression, especially via suppression of CD4+ and CD8+ T cells [64] [65] [66] [67]. Different reports have defined IL-10 producing B cells subsets, referred to as regulatory B cells. While Xiao et al. described a predominant role for PD-1-expressing B cells in IL-10 production, other reports found TIM-1+ or FcγRII low/- B cells induced by semimature DCs to be the major source of B cell IL-10 production [65] [67] [68]. Interestingly, Shalapour et al. reported IgA+ memory B cells and plasma cells in the liver as sources for IL-10 mediated suppression of CD8+ T cell immunity [69]. Two other reports highlight that the interaction between CXCR3+ cells and TAMs results in M2 macrophage polarization as well as differentiation of IgG+ plasma cells and subsequent macrophage-mediated suppression of CD8+ T cells [63] [70]. So far, it remains unclear whether all IL-10 producing and immunosuppressive B cell subsets represent distinct or rather overlapping subsets and which TME signals are responsible for driving the differentiation of B cells into either a more immunosuppressive tumor promoting or a tumor controlling phenotype. Moreover, the role of B cell antigen recognition in this context remains unclear.
Only recently, a previously unrecognized interaction between immunosuppressive B cells and dysfunctional CD8+ T cells involving the HLA-E:CD94/NKG2A axis in metastatic HCC was described [71]. Co-culture of HLA-E+ tumor infiltrating B cells with tumor infiltrating CD8+ T cells resulted in reduced CD8+ T cell functionality. Hence, targeting the NKG2A-checkpoint might be a rational approach to disrupt this immunosuppressive axis in HCC patients [71].
In sum, data regarding the role of B cells in HCC still remains controversial with both, immunosuppressive or antitumor functions, being described by different groups. Further studies are needed to fully dissect involvement of B cells in the TME in HCC, their interactions with other immune cells and their potential role in immunotherapy.
Innate and innate-like lymphocytes represent antitumor effector cells in HCC
Innate lymphoid cells (ILC) are a heterogeneous group of lymphocytes that derive from the common lymphoid progenitor but do not exhibit a rearranged antigen receptor, including NK cells. Further, innate-like T cells such as mucosal associated invariant T cells (MAIT cells) express a semi-variant TCR and recognize different classes of antigens based on molecular patterns, and may recognize tumor cells without need for prior priming with a specific antigen. In human liver, innate and innate-like lymphocytes are enriched compared to peripheral blood indicating their importance in liver immunology in general and for HCC in particular [72] [73] [74]. NK cells are traditionally considered as a key innate cell population mediating tumor immune surveillance due to their ability to recognize tumor cells using multiple germline-encoded receptor systems suitable to identify HLA-deficient and stressed cells. They can directly kill cancer cells and are also potent cytokine producers. Infiltration of NK cells is associated with improved survival [75] [76] as well as reduced HCC recurrence [77]. In animal models, unleashed NK cells were able to control HCC formation [78]. An altered NK cell phenotype with increased expression of inhibitory receptors such as the checkpoint molecule TIGIT and reduced effector functions have been described by various groups [79] [80] [81] [82]. Interestingly, TIGIT+ NK cells have also been associated with impaired control of circulating tumor cells. However, control of potential metastasis could be restored by ICI therapy targeting TIGIT [83]. Different types of NK cells are recognized that may be altered in HCC. Expanded adaptive NK cell subsets with inherently reduced effector functions were identified in patients with hepatitis B virus (HBV)-associated HCC [84]. NK cells are also affected by metabolic reprogramming in mouse models of HCC and metabolic reinvigoration of NK cells by antagonizing the secondary bile-acid iso-LCA not only partially restored NK cell intrinsic antitumor function, but also increased response to ICI therapy in mice [85]. Whether NK cells also directly respond to ICI therapy remains unclear. PD-1 expression was described on NK cells [86], however, other studies did not find significant PD-1 expression on NK cells in patients with HCC [84]. The involvement of NK cells ICI response in HCC patients requires further investigation.
MAIT cells are innate-like T cells recognizing especially non-peptide antigens derived from e.g. bacteria via their MR1-receptor [87]. MAIT cells are functionally impaired in human HCC but can be targeted by ICI therapies [88] [89]. MAIT cells were found to be restrained from infiltrating HCC tumors and functionally impaired by interaction with the immunosuppressive TME, particularly PD-L1+ TAMs. After blocking the PD-1/PD-L1 axis in murine HCC models, MAIT cell infiltration into the tumor and MAIT cell functionality was increased. ICI therapy thus targets not only classical CD8+ T cells, but also MAIT cells [89]. The exact interplay between MAIT cells and other (T) cells as targets for ICI remains, however, incompletely understood.
CD4+ T cells: A heterogeneous population with various effects on HCC
T cells represent the major target population of ICIs. CD4+ T cells coordinate the inflammatory response, adaptive immunity and CD8+ T cell function in the TME. CD4+ T helper (Th) cells can differentiate into various functionally specialized subpopulations, such as Th1, Th2, Th17, T follicular helper cells (Tfh), regulatory T cells (Tregs), and follicular regulatory T cells (Tfr) cells or cytotoxic Th cells, among others [90]. Th polarization can be distinguished based on their respective transcriptional profile and the produced cytokines. Th1 cells provide help for CD8+ T cells and govern the inflammatory type 1 response by producing large amounts of IL-2, IFN-γ and TNF-α, which was associated with an increased lymphocyte recruitment in the past [91]. Th2 cells on the other hand control type 2 responses characterized by higher amounts of IL-4 and IL-5 and a shift towards Th2 response with downregulation of Th1 response has been associated with metastasis and thus outcome of HCC [92].
A reduction of CD4+ T cells due to lipid accumulation was associated with HCC development in a mouse model of metabolic dysfunction associated steatotic liver disease (MASLD) [93] pointing towards a critical role of CD4+ T cells in the HCC formation in MASLD. An unexpected role for T cells expressing choline acetyltransferase (ChAT), the rate limiting enzyme for acetylcholine production, was recently described. Indeed, deletion of ChAT resulted in increased immunosuppression of Tregs and a dysfunction of ChAT+ conventional T cells, which favored liver cancer development [94].
The differential contribution of the different CD4+ T cell polarizations on HCC development and control, however, remains poorly understood. Existing data suggests a preferential enrichment of Tfh and Tfreg cells in HCC tissue [95]. Tfh cells are characterized by expression of CXCR5 and provide help for B cell especially in lymph nodes and the respective germinal centers [96]. Interestingly, clonal expansion of intratumoral Tfh-like CXCL13+ CD4+ T cells was associated with response to ICI therapy in another recent study [97]. Those Tfh-like CXCL13+ CD4+ cells were forming close contacts to progenitor-exhausted CD8+ T cells and specific dendritic cell populations in the TME, suggesting a regulation of progenitor-exhausted CD8+ T cells by Tfh-like CD4+ T cells and dendritic cells.
The role of regulatory T cells (Tregs) in patients with HCC has been studied by multiple groups. Tregs have multiple regulatory functions and are central mediators of immunological tolerance to autoimmunity and overwhelming inflammation. Tumors can exploit this fundamental mechanism of immune regulation to sustain tumor cell growth via induction of Tregs [98]. Tregs express the immune checkpoints CTLA-4 and PD-1, receptors targeted by current ICI therapies. Tregs can exert their regulatory role through multiple pathways, such as the production of the immunosuppressive cytokines (i.e. IL-10 and TGF-β), by outcompeting other T cells for IL-2 through expression of the high-affinity interleukin-2 receptor, by restraining costimulatory B7 molecules via CTLA-4, by inhibitory direct cell-cell contact or via promotion of local immunosuppressive adenosine signaling [99]. Thus, they can impair proliferation, activation and effector function especially of CD8+ T cells, but also other immune cells [100]. In patients with HCC, accumulation of Tregs is associated with a reduced survival and early recurrence [101] [102] [103]. Another report unveiled the role of neutrophil- derived extracellular traps in promoting Treg activity via metabolic reprogramming which resulted in an impairment in oxidative phosphorylation of Tregs and increased Treg activity [104]. In HBV-associated HCC, where Tregs are particularly enriched [102], highly immunosuppressive CCR4+ Tregs with stem-like properties were found to be attracted by tumor-derived CCL22. This promoted portal vein tumor thrombosis [105] and was associated with a poor outcome of HCC [106] [107]. Notably, in HBV-associated HCC, a pronounced epigenetic imprint of HBV-HCC on HBV-specific Tregs with high expression of e.g. HAVCR2 and PDCD1, the genes encoding the immune checkpoints Tim-3 and PD-1, was reported [108]. In summary, Tregs represent a major T cell population driving an immunosuppressive TME in patients with HCC.
CD8+ T cells are primary effector cells in HCC under ICI therapy
CD8+ T cells represent a major effector arm of the adaptive immune system. Naive conventional CD8 + T cells can recognize peptide antigens presented via MHC class I molecules on specialized antigen presenting cells such as dendritic cells or macrophages via their TCR. For complete activation and induction of a strong effector and memory response, T cells integrate 4 major signals, the TCR signal, costimulatory signals like CD28 and cytokines as well as metabolic cues [109]. Effector functions with antiviral or antitumor properties comprise both the release of various cytokines, but also direct cytotoxic effects mediated through cytotoxic molecules like perforin and granzymes or death receptors [110] [111]. Thus, CD8+ T cells can differentiate into effector T cells with potent antitumor functions. In patients with HCC, high CD8+ T cell infiltration is associated with a favorable prognosis [95] [102] [112] [113] [114]. However, in HCC similar to many other cancers and viral infection, persistent antigen and inflammatory and immunoregulatory cues may induce a program of T cell exhaustion. T cell exhaustion represents a T cell program associated with reduced effector function adapted to chronic stimulation. It is characterized by an increased expression of inhibitory receptors, a dysfunctional effector program as well as transcriptional, epigenetic and metabolic alterations [115] (Infobox 1). Notably, PD-1 is a central regulator of the metabolic and proliferative program of exhausted cells [116] and exhausted T cells represent the central correlate of immune reinvigoration after ICI therapy [117] [118] ([Fig. 4]).

T cell exhaustion was first described in the context of chronic viral infections in mouse and men [119] [120], but is also frequent in multiple human cancers [121]. Upon chronic antigen stimulation especially in inflammatory conditions, exhausted T cells (Tex) progressively loose effector functions (e.g. reduced cytokine production and direct cytotoxicity) and upregulate inhibitory receptors such as PD-1, Tim-3, LAG-3 and others [115]. Tex display a profoundly altered transcriptional, epigenetic and metabolic program compared to effector or memory T cells [116] [122] [123]. These alterations can be considered as suppressed cell states, however, Tex are now more plausibly perceived as adapting to the inflammatory milieu and preserving residual effector functions to maintain immune responses [109] [124] [125]. It has become clear that Tex are heterogeneous and developmental hierarchies exist within the exhausted cell program. Progenitor Tex (Tpex) have enhanced self-renewal, metabolic fitness and high proliferative capacity and express the transcription factor TCF-1 that can give rise to terminal exhausted T cells (Ttex) that have high expression of TOX, Eomes and dysfunctional cytokine production [126] [127] [128]. Of note, the Tpex population represents the primary CD8+ T cell population responding to ICI therapy and clinical response of Tpex frequency may be associated with the immunological response of Tpex [129] [130] [131] [132].
T cell exhaustion and tissue resident memory T cells – related in phenotype, separated in function?
Exhausted T cells (Tex) (Infobox 1), often profiled by their high expression of PD-1 and exhibiting limited effector function, have been characterized in HCC patients and in HCC tumor tissue with an enrichment of Tex in tumor-infiltrating lymphocytes (TILs) [95] [133] [134] [135] [136] [137]. Consistently, single cell RNA sequencing revealed an upregulation of CD8+ T cells with high expression of inhibitory receptors like PD-1, CTLA-4 or TIGIT (T-cell immunoreceptor with Ig and ITIM domains) [138] and a high infiltration of Tex was associated with impaired survival of HCC patients [135] [139] [140] [141]. Tex in HCC were further reported to be induced by tumor associated methionine metabolism, which was connected to reduced chromatin accessibility in activated T cells [139]. However, a clear differentiation of Tex solely by expression of inhibitory receptors in general and PD-1 in particular, is misleading, as PD-1 is not only a marker of exhausted T cells, but also present e.g. on activated T cells [141], and subsets of memory T cells, especially tissue resident memory T cells (Trm) [95]. Trm express markers associated with tissue homing and residency but this program overlaps with exhausted cells. Of note, PD-1 is also highly present on Progenitor Tex (Tpex) [142]. Thus, for a thorough understanding of T cell subsets in the TME, a comprehensive profiling of phenotypic and ideally functional features is required. The use of high-dimensional single cell proteomics in patients with HCC revealed the presence of Trm cells that were characterized by coexpression of PD-1 with markers of tissue resident T cells, especially CD103. These Trm displayed fewer coexpression of other inhibitory receptors and transcription factors associated with Tex and an overall increased functionality and metabolism compared to bona fide Tex. Importantly, high infiltration of Trm was associated with a good prognosis in patients with HCC [95]. Thus, the balance of exhausted vs tissue resident memory T cells may have prognostic potential in HCC. Other studies support the protective function of Trm in patients with HCC [143] [144] [145]. In patients with HBV-associated HCC, high infiltration of CD69+ CD103+ Trm targeting HBV-epitopes was associated with a reduction of HCC relapse. Of note, these Trm lacked features of terminally exhaustion while expressing intermediate levels of PD-1 [133]. Recently, single cell RNA sequencing revealed a pronounced heterogeneity in Trm cells in patients with HCC in terms of involved cytokine networks [144]. However, most of the evidence underpinning the positive prognostic value of Trm in patients with HCC relies on HBV-associated HCC, which may not be representative for other tumor etiologies. Indeed, PD-1+ CD8+ T cells with increased expression of the tissue-residency marker CXCR6, thus considered to be Trm cells, were associated with an increased liver pathology and subsequent HCC development rather than with tumor control in a mouse model of MASLD-associated HCC [23]. Another report revealed in a mouse model mimicking MASLD, that hepatic CXCR6+ PD-1+ CD8+ T cells can become auto-aggressive following alternative, MHC class I independent activation in an inflammatory, metabolically disturbed microenvironment in livers with MASLD [146]. In the future, it will thus be important to further dissect the role of CD8+ T Trm and Tex in patients with HCC considering both the involvement of different HCC etiologies on T cell differentiation and exhaustion as well T cell heterogeneity in the TME in order to maximize treatment efficacy of ICI therapy.
Role of tumor antigens in HCC
Another unresolved yet translationally highly important question is which antigens are targeted by CD8+ T cells in the HCC TME, and how antigen specificity impacts antitumor function of CD8+ T cells as well as response to ICI. Epitopes potentially targeted by CD8+ T cells in the HCC TME include different groups of antigens: viral antigen, tumor-associated antigens as well as mutation-associated epitopes (neoantigens) [114]. For patients with virus-associated HCC, viral antigens might still be present in HCC and represent a potential target for CD8+ T cells. In line with this, HBV-specific CD8+ T cells were highly present in the HCC tumor tissue of patients with HBV-associated HCC. These HBV-specific CD8+ T cells lacked features of terminal exhaustion and were associated with a better prognosis of HBV-associated HCC [133]. Tumor-associated antigens (TAAs) represent germline-encoded (unmutated) antigens highly expressed in tumor cells, such as cancer testis antigens, whose expression in non-cancerous tissue is limited to the testes or placenta [147]. AFP is a prominent TAA in HCC. TAAs presented by MHC molecules can be readily detected in HLA ligandomes from HCC patients [148] [149] and presence of TAA-specific CD8+ T cells was associated with an increased progression-free survival in patients with HCC [150]. TAA-specific CD8+ T cells have also been reported to be dysfunctional with a concomitant upregulation of inhibitory receptors targetable by ICIs such as PD-1 or LAG-3 [151]. ICI blockade can increase TAA-specific CD8+ T cell responses [152]. Nevertheless it is unclear if TAA-specific CD8+ T cells indeed recognize tumors and undergo exhaustion in patients [152]. Thus, the extent to which TAA-specific CD8+ T cells contribute to clinical response to ICI therapy remains currently unclear. Finally, neoantigens, i.e. tumor-specific antigens resulting mostly from tumor intrinsic mutations represent potentially relevant CD8+ T cell antigens. Current evidence from other tumors apart from HCC suggests, that neoantigen-specific CD8+ T cells are the primary effector cells responding to ICIs [153]. A thorough understanding of neoantigen specific CD8+ T cells as targets for immunotherapy in HCC however, is still an unmet scientific need.
Detection and identification of neonantigens and particularly of neoantigen-specific CD8+ T cells is challenging as neoantigens are usually patient-specific and require careful genomic and immunologic analysis [154]. Moreover, in HCC patients, considerable heterogeneity of neoantigens may exist between different tumor nodules [148]. This intrapatient heterogeneity was also observed for metastatic HCC nodules, where high neoantigen hetereogeneity was associated with impaired T-cell mediated immunity and a shortened overall survival [71]. As most studies used computational or transcriptome based approaches to identify neoantigens in patients with HCC [155], experimental proof of immunogenicity is scarce [156]. One study identified CD39+ CD8+ T cells as a surrogate for the CD8+ T cell population recognizing neoantigens with high-affinity after presentation by dendritic cells. Interestingly, higher amounts of high-affinity neoantigens on the one hand and CD39+ CD8+ T cells among TILs (tumor infiltrating lymphocytes) on the other hand was found to be associated with a prolonged overall survival [157]. Whether all neonantigen-specific CD8+T cells invade the tumor, is another insufficiently understood subject. Maravelia and colleagues recently highlighted differences between neoantigen-specific CD8+ T cells identified in tumor draining lymph nodes with features of tissue residency and exhaustion, whereas the neoantigen-specific CD8+ T cell population from liver flushes had characteristics of effector memory cells [158]. While neoantigen-specific T cell responses hold promise for improving HCC therapy, mechanisms of tumor immune escape may impair (neo-)antigen presentation to (CD8+) T cells by tumor cells, such as HLA loss of heterozygosity [71] [148]. More research in HCC is required to understand if neoantigen-specific responses are central to the effect of ICI therapy.
Another interesting strategy targeting neoantigen-specific CD8+ T cells in HCC is neoantigen-based vaccination, which has been already successfully applied in early clinical trials for pancreatic cancer [159]. Recently, a first early phase clinical trial evaluated immunogenitcity and safety of a personalized therapeutic cancer vaccine (PTCV) based on individually selected neoantigens in combination with PD-1 blockade using pembrolizumab in patients with advanced HCC previously treated with multikinase inhibitors [160]. Neoantigens were identified using DNA and RNA sequencing and used as an individual vaccine for patients with advanced HCC after being encoded in a plasmid. Out of the 36 patients included, the objective response rate was 30.6% with 8.3% of the patients achieving a complete response. Immunologically, PTCV in combination with ICI lead to induction of new PTCV-specific T cell responses displaying predominantly a CD8+ effector memory phenotype as well as expansion of preexisting responses [160]. Future studies including control groups will be needed to fully understand the therapeutic potential of PTCV (in combination with ICI) as a treatment for patients with HCC and to develop surrogate markers for patients responding to PTCV.
Endothelial cells control immune cell infiltration and contribute to an immunosuppressive milieu
Endothelial cells play an important role in cancer biology. The liver endothelial cell compartment includes macrovascular endothelial cells and liver sinusoidal endothelial cells (LSECs). Particularly LSECs are thought to impact on the composition and phenotype of the TME in HCC by controlling blood flow and immune cell filtration. Thus, tumor associated endothelial cells (TEC’s) of the liver show an altered structural and functional phenotype with perturbed fenestration, augmented angiogenic capacity and immunosuppressive interactions [161]. Indeed, recent studies involving single cell RNA sequencing and spatial transcriptomics revealed distinct molecular signatures that differentiate TEC’s from normal LSECs. Zhou et al report TGFβ-signaling to drive an immunosuppressive TEC phenotype [162], while Xie et al. suggest a role of Notch signaling for TEC differentiation in HCC [163]. TECs hereby typically overexpress plasmalemma vesicle associated protein (PLVAP) that contributes to the altered vascular permeability in tumor vessels [164]. Functionally, TECs particularly attract regulatory T cells (Tregs) via specific adhesion molecules such as VCAM-1 and CD151 as well as chemokines (CXCL10, CCL2, CCL3) while rather repressing infiltration of effector cells, like dendritic cells or NK cells [165]. Besides its role as selective gatekeepers, a distinct subset of TECs actively impairs CD8+ T cell differentiation e.g. by secretion of CXCL12 [166]. TECs can further mediate classical immunological functions, such as antigen presentation. However, in contrast to other APC’s, antigen presentation by TECs contributes to CD8 T cell tolerance [167] [168]. Finally, the abnormal, leaky tumor blood vessels shaped by TECs drive a hypoxic and nutrient-deprived microenvironment, suppressing T cell activity [169]. Overall, TEC’s in HCC foster tumor immune evasion by shaping an immunosuppressive tumor environment and are therefore attractive therapeutic targets. Indeed, antiangiogenic therapies targeting VEGF signaling (e.g. Bevacizumab but also TKI) have shown efficacy in HCC and can mediate tumor-suppressive effects by reducing tumor blood supply and limiting neoangiogenesis, thereby impacting on the TME. Specifically, Bevacizumab treatment was associated with increased infiltration of immune cells, particularly cytotoxic CD8+ T cells, and bevacizumab combined with ICI has shown synergistic effects on immune cell infiltration in preclinical studies [170] [171]. However, VEGF signaling is not restricted to endothelial cells, suggesting that the immunomodulatory effects of bevacizumab or TKI’s might not be exclusively mediated by normalization of tumor vasculature.
Stromal cells impair tumor immune control by balancing the TME towards immunosuppressive subpopulations
As HCC typically arises in fibrotic livers, malignant cells are often surrounded by diverse mesenchymal cells and abundant extracellular matrix (ECM). In addition to activated hepatic stellate cells differentiating into myofibroblasts, the predominant stromal cell type in healthy liver, mesenchymal cell types in HCC include Cancer-associated Fibroblasts (CAFs) and Mesenchymal Stem Cells (MSC) [172]. Particularly CAFs play a critical role in HCC progression by promoting tumor cell proliferation and by modulating immune responses. In fact, transcriptomic signatures of CAFs are linked to aggressive tumor behavior and poor prognosis in HCC [173]. CAFs originate from diverse progenitors, primarily hepatic stellate cells (HSC), but also portal (PF) and vascular smooth muscle cells [174]. Recent single-cell RNA sequencing (scRNA-seq) studies in HCC suggest at least 3 distinct CAF subpopulations in HCC with transcriptomic signatures that reflect its potential cellular origin: RGS5high, THY1high HSC_CAFs, MYH11high VSMC_CAFs and PDGFRA, MMP23Bhigh PF_CAFs [175]. Compared to physiological fibroblasts, CAFs show upregulation of tumor-promoting cytokines and growth factors, such as CXCL6, PDGF-AB, osteopontin and HGF, and may thereby drive tumor cell survival, migration, stemness and drug resistance [21]. CAFs can also shape the tumor immune microenvironment and promote development of an immune cell excluded or immunosuppressive tumor niche. Thus, CAFs may limit immune cell infiltration into the tumor by production of an ECM that acts as a physical barrier and hinders immune cell infiltration. Moreover, the ECM controls spatial distribution and bioavailibility of cytokines and growth factors. Release of these factors by proteolytic enzymes strongly influences immune cell recruitment, maintenance and polarization. Particularly, persistence of immunosuppressive cell populations, such as Tregs, are supported by ECM-components of HCC, such as hyaluronan [176]. CAFs can further produce immunosuppressive cytokines and growth factors itself. Hereby, particularly TGF-β appears to play a key role. Thus, CAF-derived TGF-β contributes to exclusion of cytotoxic T cells [177] and impairs T cell differentiation and activation [178]. In fact, TGF-β suppresses expression of key effector molecules such as perforin, granzymes and IFN-γ, limiting the cytotoxicity of CD8+ T cells [179]. Finally, CAFs are linked to enhanced recruitment of immunosuppressive myeloid cells. Thus, CD36+ HSC_CAFs have been shown to recruit CD33+ MDSC’s via secretion of macrophage migration inhibitory factor (MIF) [180]. Endosialin expression in CAFs on the other hand is associated with macrophage M2 polarization [174].
Besides CAFs, the stromal compartment in HCC includes MSCs that may be recruited to the tumor site via chemokines such as IL-8 or MCP1 secreted by tumor cells [181]. MSCs are less well studied in HCC compared to CAFs but seem to play a dual role with both immunosuppressive and immune-activating functions being reported. On the one hand, MSCs have been reported to attract CD8 T cells [182] and to inhibit tumor growth by increasing the infiltration of granulocytes and monocytes [183]. On the other hand, MSCs secrete immunosuppressive cytokines, such as IL-10 or TGF-β and have been reported to inhibit dendritic cells while promoting M2 macrophage polarization [184] [185]. These data indicate that the stromal cell compartment in HCC fosters immunosuppression and therefore represents an attractive target to boost anti-tumor immune control. Unspecific targeting of fibroblasts, however, is ineffective for tumor control [186]. Nevertheless, of the mechanisms how the stroma shapes the TME requires further exploration.
Spatial analysis of the TME as predictor for checkpoint therapy responses
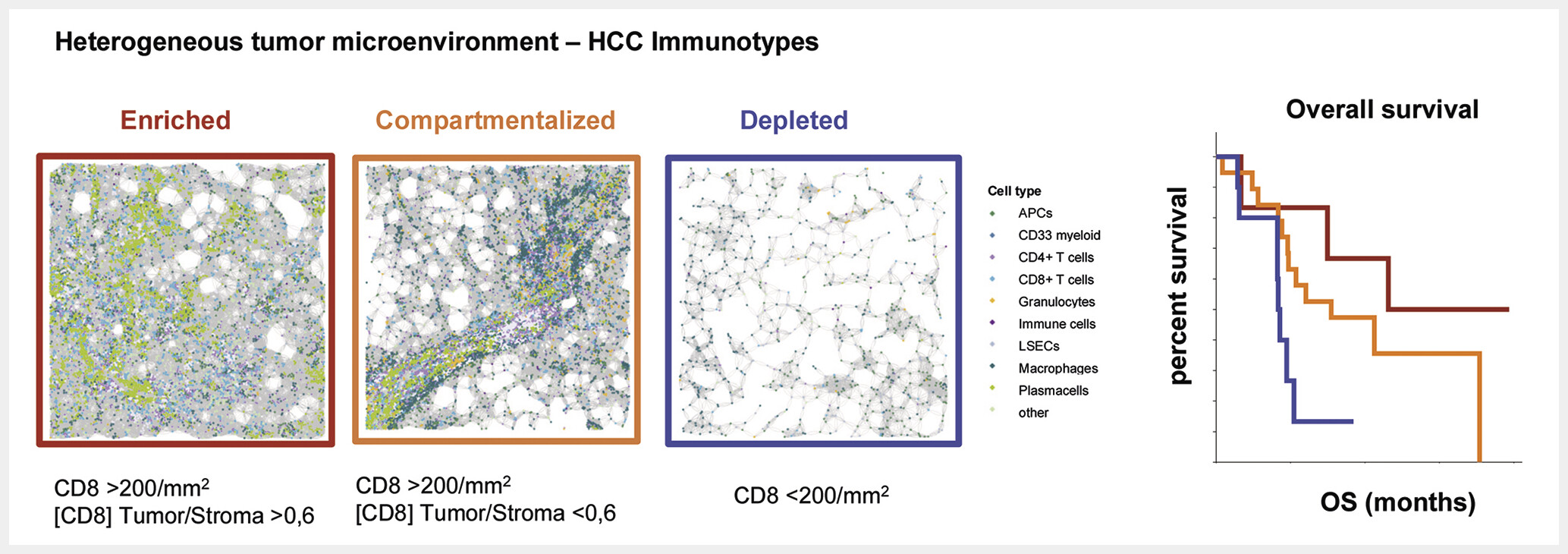
While many insights into immune cell populations in HCC have been gained from focused analysis of isolated cell populations, clear links predicting response to ICIs using specific cell populations are missing. Scores commonly used for other tumors such as the PD-L1 based CPS or TPS score perform poorly in HCC (reviewed elsewhere [138]). The etiology of liver cancer may also impact ICI responses. Especially patients with chronic viral hepatitis seem to benefit from ICI, and checkpoint therapy is described to promote procancerogenic effects in specific models of metabolic associated HCC [23] [187]. The clinical relevance of these findings requires further evaluation in prospective studies. Current guidelines do not limit ICI to HCC patients with certain underlying liver diseases [188]. Advances in spatial profiling methods now enable a comprehensive profiling of the tumor immune microenvironment. A central finding from studies analyzing large patient cohorts is that the immune infiltrate in HCC is highly heterogeneous across patients. Interacting immune cells in the tumor microenvironment that are associated with improved prognosis have been identified based on topological analysis [189]. The spatial distribution of immune infiltrates in general and NK cells in particular may predict HCC recurrence risk [77]. In patients with ICI therapy, interactions of cellular triads consisting of CD8+ Tpex, CD4+ CXCL13+ T cells around dendritic cells were associated with favorable responses and suggestive of local differentiation upon ICI therapy [97]. Other studies highlighted a role for tertiary lymphoid structures (TLS) in HCC. For example, Shu et al. recently demonstrated that CD20⁺CXCL13⁺ lymphoid aggregates – surrogate markers for TLS – can serve as prognostic biomarkers of immunotherapy response [190]. Thus, spatial immune features may predict outcome after HCC ICI therapy. In a recent analysis of the heterogeneity of the HCC TME using comprehensive spatial profiling of immune cell subsets, three major types of immune cell interactions in microanatomic niches were identified that involved different immune cell types. Importantly, one niche associated with CD8+ T cells interacting with B cells and plasma cells was associated with ICI response [191]. The work further highlighted the relevance of intratumoral immune infiltration. Indeed, three major spatial immunotypes in HCC were identified that could be defined based on the extent and distribution of infiltrating immune cells – an immune depleted class, a compartmentalized class (preferential retention of immune cells in the tumor stroma with a prominent role for myeloid cell-T cell interactions), and an enriched class (highest accumulation of CD8+ T cells in tumor parenchyma) [191]. Interestingly, a classification of these immune subtypes was possible not only by spatial omics methods but could also be achieved using CD8+ T cell infiltration as a simplified surrogate marker. Importantly, the spatial immune classification trained in an independent dataset was predictive for patient outcome in a validation cohort, with the enriched immunotype associated with the longest survival after ICI therapy [191] ([Fig. 5]). These findings are potentially practice changing since a simplified analysis of CD8+ T cell infiltration into the tumor can be performed during routine histopathological evaluation of HCC and may inform about patients that could be prioritized for ICI therapies in early stages of HCC. However, the predictive role of such biomarkers requires further evaluation in prospective studies.
Conclusion
The introduction of immunotherapies in HCC constitutes a significant advance in patient care. It is apparent that responses can be complete and durable, but many patients also fail to respond to therapy. T cell responses represent a central correlate of successful ICI therapies, however, many immunological cell types and regulatory mechanisms have been identified that shape the tumor immune microenvironment in HCC and may limit responses. Abundance or exclusion of various cellular components of the TME have been associated with ICI response. In particular, CD8+ T cell infiltration correlates with an immunoresponsive HCC phenotype. Suppressive myeloid cells on the other cells are rather enriched in immunoresistant HCC. Yet, the individual cell-type specific observations did not translate directly into clinical application as reliable biomarkers. A prominent observation is that immune response are highly heterogeneous in HCC patients. Approaches to understand the immune cell interactions that are associated with favorable prognosis using comprehensive spatial profiling have revealed central roles for interactions between different T cell and myeloid cell subsets. Moreover, novel diagnostic approaches for HCC using the immune architecture of the HCC TME to classify spatial immunotypes linked to outcome of immune checkpoint inhibitor therapy may help to inform patients that should be prioritized for ICI therapy. These data indicate that an evaluation of the immune architecture of the HCC TME should become standard in future clinical trials.
Conflict of Interest
BB has received honoraria from AstraZeneca, BMS and Roche.
-
References
- 1 Zentrum für Krebsregisterdaten RKI. Krebs in Deutschland, p 44–47. 2023 Accessed August 10, 2025 at: https://www.krebsdaten.de/Krebs/DE/Content/Publikationen/Krebs_in_Deutschland/krebs_in_deutschland_2023.pdf?__blob=publicationFile
- 2 Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 1999; 19: 329-338
- 3 Reig M, Forner A, Rimola J. et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J Hepatol 2022; 76: 681-693
- 4 Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378-390
- 5 Cheng A-L, Kang Y-K, Chen Z. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. The Lancet Oncology 2009; 10
- 6 Kudo M, Finn RS, Qin S. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 2018; 391: 1163-1173
- 7 El-Khoueiry AB, Sangro B, Yau T. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. The Lancet 2017; 389: 2492-2502
- 8 Yau T, Park JW, Finn RS. et al. LBA38_PR – CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Annals of Oncology 2019; 30: v874-v875
- 9 Zhu AX, Finn RS, Edeline J. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. The Lancet Oncology 2018; 19: 940-952
- 10 Finn RS, Ryoo B-Y, Merle P. et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2020. Accessed August 03, 2020 at: https://pubmed.ncbi.nlm.nih.gov/31790344/
- 11 Finn RS, Qin S, Ikeda M. et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. The New England journal of medicine 2020. Accessed August 03, 2020 at: https://pubmed.ncbi.nlm.nih.gov/32402160/
- 12 Abou-Alfa GK, Lau G, Kudo M. et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evidence 2022; 1: EVIDoa2100070
- 13 Yau T, Galle PR, Decaens T. et al. Nivolumab plus ipilimumab versus lenvatinib or sorafenib as first-line treatment for unresectable hepatocellular carcinoma (CheckMate 9DW): an open-label, randomised, phase 3 trial. The Lancet 2025; 405: 1851-1864
- 14 Qin S, Kudo M, Meyer T. et al. Tislelizumab vs Sorafenib as First-Line Treatment for Unresectable Hepatocellular Carcinoma: A Phase 3 Randomized Clinical Trial. JAMA oncology 2023; 9
- 15 Kelley RK, Rimassa L, Cheng A-L. et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology 2022; 23
- 16 Llovet JM, Kudo M, Merle P. et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind, phase 3 trial. The Lancet Oncology 2023; 24
- 17 Qin S, Chan SL, Gu S. et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international phase 3 study. Lancet (London, England) 2023; 402
- 18 Scheiner B, Kang B, Balcar L. et al. Outcome and management of patients with hepatocellular carcinoma who achieved a complete response to immunotherapy-based systemic therapy. Hepatology 2025; 81: 1714-1727
- 19 Horst AK, Neumann K, Diehl L. et al. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol Immunol 2016; 13: 277-292
- 20 Zheng M, Tian Z. Liver-Mediated Adaptive Immune Tolerance. Front Immunol 2019; 10
- 21 Rennert C, Lang-Meli J, Gromak M. et al. Perspectives for novel therapeutic concepts in hepatocellular carcinoma targeting the stromal and innate immune microenvironment. Liver Cancer International 2023; 4: 42-57
- 22 Calderaro J, Ziol M, Paradis V. et al. Molecular and histological correlations in liver cancer. Journal of Hepatology 2019; 71: 616-630
- 23 Pfister D, Núñez NG, Pinyol R. et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021; 592: 450-456
- 24 Sharma A, Seow JJW, Dutertre C-A. et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020; 183: 377-394.e21
- 25 Lanzanò L, Coto Hernández I, Castello M. et al. Encoding and decoding spatio-temporal information for super-resolution microscopy. Nat Commun 2015; 6: 6701
- 26 Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011; 11: 762-774
- 27 Yang F, Wei Y, Han D. et al. Interaction with CD68 and Regulation of GAS6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res 2020; 80: 3892-3905
- 28 Yuan Y, Wu D, Li J. et al. Mechanisms of tumor-associated macrophages affecting the progression of hepatocellular carcinoma. Front Pharmacol 2023; 14
- 29 Liu P-S, Wang H, Li X. et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017; 18: 985-994
- 30 Tripathi C, Tewari BN, Kanchan RK. et al. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5: 5350-5368
- 31 Lu Y, Yang A, Quan C. et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun 2022; 13: 4594
- 32 Fan G, Xie T, Li L. et al. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol 2024; 8: 75
- 33 Tan J, Fan W, Liu T. et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J Hepatol 2023; 79: 126-140
- 34 Zhou L, Wang M, Guo H. et al. Integrated Analysis Highlights the Immunosuppressive Role of TREM2+ Macrophages in Hepatocellular Carcinoma. Front Immunol 2022; 13: 848367
- 35 Roderfeld M, Rath T, Lammert F. et al. Innovative immunohistochemistry identifies MMP-9 expressing macrophages at the invasive front of murine HCC. World J Hepatol 2010; 2: 175-179
- 36 Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 2011; 32: 19-25
- 37 Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res 2017; 5: 3-8
- 38 Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017; 8: 3649-3665
- 39 Mandruzzato S, Brandau S, Britten CM. et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother 2016; 65: 161-169
- 40 Bronte V, Brandau S, Chen S-H. et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7: 12150
- 41 Hoechst B, Ormandy LA, Ballmaier M. et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008; 135: 234-243
- 42 Arihara F, Mizukoshi E, Kitahara M. et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother 2013; 62: 1421-1430
- 43 Zhang X, Fu X, Li T. et al. The prognostic value of myeloid derived suppressor cell level in hepatocellular carcinoma: A systematic review and meta-analysis. PLoS One 2019; 14: e0225327
- 44 Ma C, Zhang Q, Greten TF. MDSCs in liver cancer: A critical tumor-promoting player and a potential therapeutic target. Cell Immunol 2021; 361: 104295
- 45 Chiu DK-C, Xu IM-J, Lai RK-H. et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016; 64: 797-813
- 46 Kasic T, Colombo P, Soldani C. et al. Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol 2011; 41: 1843-1849
- 47 Kusmartsev S, Nefedova Y, Yoder D. et al. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol 2004; 172: 989-999
- 48 Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol 2018; 9: 2499
- 49 Hoechst B, Voigtlaender T, Ormandy L. et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009; 50: 799-807
- 50 Lacotte S, Slits F, Orci LA. et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology 2016; 5: e1234565
- 51 Tomiyama T, Itoh S, Iseda N. et al. Myeloid‑derived suppressor cell infiltration is associated with a poor prognosis in patients with hepatocellular carcinoma. Oncology Letters 2022; 23: 1-9
- 52 Del Prete A, Salvi V, Soriani A. et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol 2023; 20: 432-447
- 53 Ormandy LA, Färber A, Cantz T. et al. Direct ex vivo analysis of dendritic cells in patients with hepatocellular carcinoma. World J Gastroenterol 2006; 12: 3275-3282
- 54 Saito Y, Komori S, Kotani T. et al. The Role of Type-2 Conventional Dendritic Cells in the Regulation of Tumor Immunity. Cancers (Basel) 2022; 14: 1976
- 55 Li W, Chen G, Peng H. et al. Research Progress on Dendritic Cells in Hepatocellular Carcinoma Immune Microenvironments. Biomolecules 2024; 14: 1161
- 56 Suthen S, Lim CJ, Nguyen PHD. et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology 2022;
- 57 Pang L, Yeung OWH, Ng KTP. et al. Postoperative Plasmacytoid Dendritic Cells Secrete IFNα to Promote Recruitment of Myeloid-Derived Suppressor Cells and Drive Hepatocellular Carcinoma Recurrence. Cancer Res 2022; 82: 4206-4218
- 58 Shi J-Y, Gao Q, Wang Z-C. et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin Cancer Res 2013; 19: 5994-6005
- 59 Brunner SM, Itzel T, Rubner C. et al. Tumor-infiltrating B cells producing antitumor active immunoglobulins in resected HCC prolong patient survival. Oncotarget 2017; 8: 71002-71011
- 60 Garnelo M, Tan A, Her Z. et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017; 66: 342-351
- 61 Trailin A, Červenková L, Ambrozkiewicz F. et al. T- and B-Cells in the Inner Invasive Margin of Hepatocellular Carcinoma after Resection Associate with Favorable Prognosis. Cancers (Basel) 2022; 14: 604
- 62 Faggioli F, Palagano E, Di Tommaso L. et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018; 67: 1970-1985
- 63 Wei Y, Lao X-M, Xiao X. et al. Plasma Cell Polarization to the Immunoglobulin G Phenotype in Hepatocellular Carcinomas Involves Epigenetic Alterations and Promotes Hepatoma Progression in Mice. Gastroenterology 2019; 156: 1890-1904.e16
- 64 Neo SY, Shuen TWH, Khare S. et al. Atypical memory B cells acquire Breg phenotypes in hepatocellular carcinoma. JCI Insight 2025; 10: e187025
- 65 Ouyang F-Z, Wu R-Q, Wei Y. et al. Dendritic cell-elicited B-cell activation fosters immune privilege via IL-10 signals in hepatocellular carcinoma. Nat Commun 2016; 7: 13453
- 66 Shao Y, Lo CM, Ling CC. et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett 2014; 355: 264-272
- 67 Xue H, Lin F, Tan H. et al. Overrepresentation of IL-10-Expressing B Cells Suppresses Cytotoxic CD4+ T Cell Activity in HBV-Induced Hepatocellular Carcinoma. PLoS One 2016; 11: e0154815
- 68 Xiao X, Lao X-M, Chen M-M. et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov 2016; 6: 546-559
- 69 Shalapour S, Lin X-J, Bastian IN. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017; 551: 340-345
- 70 Liu R-X, Wei Y, Zeng Q-H. et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology 2015; 62: 1779-1790
- 71 Sun Y, Wu P, Zhang Z. et al. Integrated multi-omics profiling to dissect the spatiotemporal evolution of metastatic hepatocellular carcinoma. Cancer Cell 2024; 42: 135-156.e17
- 72 Dusseaux M, Martin E, Serriari N. et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17–secreting T cells. Blood 2011; 117: 1250-1259
- 73 Heymann F, Tacke F. Immunology in the liver — from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016; 13: 88-110
- 74 Hudspeth K, Donadon M, Cimino M. et al. Human liver-resident CD56bright/CD16neg NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J Autoimmun 2016; 66: 40-50
- 75 Taketomi A, Shimada M, Shirabe K. et al. Natural killer cell activity in patients with hepatocellular carcinoma: a new prognostic indicator after hepatectomy. Cancer 1998; 83: 58-63
- 76 Xue J-S, Ding Z-N, Meng G-X. et al. The Prognostic Value of Natural Killer Cells and Their Receptors/Ligands in Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. Front Immunol 2022; 13: 872353
- 77 Jia G, He P, Dai T. et al. Spatial immune scoring system predicts hepatocellular carcinoma recurrence. Nature 2025; 640: 1031-1041
- 78 Molgora M, Bonavita E, Ponzetta A. et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017; 551: 110-114
- 79 Cai L, Zhang Z, Zhou L. et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol 2008; 129: 428-437
- 80 Sun H, Huang Q, Huang M. et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019; 70: 168-183
- 81 Yoshida Y, Yoshio S, Yamazoe T. et al. Phenotypic Characterization by Single-Cell Mass Cytometry of Human Intrahepatic and Peripheral NK Cells in Patients with Hepatocellular Carcinoma. Cells 2021; 10: 1495
- 82 Yu L, Liu X, Wang X. et al. TIGIT+ TIM-3+ NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus‑related hepatocellular carcinoma. Oncoimmunology 2021; 10: 1942673
- 83 Sun Y, Li T, Ding L. et al. Platelet-mediated circulating tumor cell evasion from natural killer cell killing through immune checkpoint CD155-TIGIT. Hepatology 2025; 81: 791-807
- 84 Rennert C, Tauber C, Fehrenbach P. et al. Adaptive Subsets Limit the Anti-Tumoral NK-Cell Activity in Hepatocellular Carcinoma. Cells 2021; 10: 1369
- 85 Wei H, Suo C, Gu X. et al. AKR1D1 suppresses liver cancer progression by promoting bile acid metabolism-mediated NK cell cytotoxicity. Cell Metab 2025; 37: 1103-1118.e7
- 86 Pesce S, Greppi M, Tabellini G. et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol 2017; 139: 335-346.e3
- 87 Treiner E, Duban L, Bahram S. et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003; 422: 164-169
- 88 Duan M, Goswami S, Shi J-Y. et al. Activated and Exhausted MAIT Cells Foster Disease Progression and Indicate Poor Outcome in Hepatocellular Carcinoma. Clinical Cancer Research 2019; 25: 3304-3316
- 89 Ruf B, Bruhns M, Babaei S. et al. Tumor-associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell 2023; 186: 3686-3705.e32
- 90 Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010; 28: 445-489
- 91 Hirano S, Iwashita Y, Sasaki A. et al. Increased mRNA expression of chemokines in hepatocellular carcinoma with tumor-infiltrating lymphocytes. Journal of Gastroenterology and Hepatology 2007; 22: 690-696
- 92 Budhu A, Forgues M, Ye Q-H. et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006; 10: 99-111
- 93 Ma C, Kesarwala AH, Eggert T. et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016; 531: 253-257
- 94 Zheng C, Snow BE, Elia AJ. et al. Tumor-specific cholinergic CD4+ T lymphocytes guide immunosurveillance of hepatocellular carcinoma. Nat Cancer 2023; 4: 1437-1454
- 95 Barsch M, Salié H, Schlaak AE. et al. T cell exhaustion and residency dynamics inform clinical outcomes in hepatocellular carcinoma. J Hepatol 2022;
- 96 Deenick EK, Ma CS. The regulation and role of T follicular helper cells in immunity. Immunology 2011; 134: 361-367
- 97 Magen A, Hamon P, Fiaschi N. et al. Intratumoral dendritic cell-CD4+ T helper cell niches enable CD8+ T cell differentiation following PD-1 blockade in hepatocellular carcinoma. Nat Med 2023; 29: 1389-1399
- 98 Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012; 72: 2162-2171
- 99 Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell 2023; 41: 450-465
- 100 Schmidt A, Oberle N, Krammer PH. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front Immunol 2012; 3
- 101 Huang Y, Liao H, Zhang Y. et al. Prognostic Value of Tumor-Infiltrating FoxP3+ T Cells in Gastrointestinal Cancers: A Meta Analysis. PLoS ONE 2014; 9: e94376
- 102 Lim CJ, Lee YH, Pan L. et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019; 68: 916-927
- 103 Tu J-F, Ding Y-H, Ying X-H. et al. Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep 2016; 6: 35056
- 104 Wang H, Zhang H, Wang Y. et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol 2021; 75: 1271-1283
- 105 Yang P, Li Q-J, Feng Y. et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012; 22: 291-303
- 106 Llovet JM, Bustamante J, Castells A. et al. Natural history of untreated nonsurgical hepatocellular carcinoma: rationale for the design and evaluation of therapeutic trials. Hepatology 1999; 29: 62-67
- 107 Schöniger-Hekele M, Müller C, Kutilek M. et al. Hepatocellular carcinoma in Central Europe: prognostic features and survival. Gut 2001; 48: 103-109
- 108 You M, Gao Y, Fu J. et al. Epigenetic regulation of HBV-specific tumor-infiltrating T cells in HBV-related HCC. Hepatology 2023; 78: 943-958
- 109 Giles JR, Globig A-M, Kaech SM. et al. CD8+ T cells in the cancer-immunity cycle. Immunity 2023; 56: 2231-2253
- 110 Barry M, Bleackley RC. Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2002; 2: 401-409
- 111 Koh C-H, Lee S, Kwak M. et al. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp Mol Med 2023; 55: 2287-2299
- 112 Gabrielson A, Wu Y, Wang H. et al. Intratumoral CD3 and CD8 T-cell Densities Associated with Relapse-Free Survival in HCC. Cancer Immunol Res 2016; 4: 419-430
- 113 Gao Q, Qiu S-J, Fan J. et al. Intratumoral Balance of Regulatory and Cytotoxic T Cells Is Associated With Prognosis of Hepatocellular Carcinoma After Resection. JCO 2007; 25: 2586-2593
- 114 Hofmann M, Tauber C, Hensel N. et al. CD8+ T Cell Responses during HCV Infection and HCC. J Clin Med 2021; 10: 991
- 115 McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 2019; 37: 457-495
- 116 Bengsch B, Johnson AL, Kurachi M. et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016; 45: 358-373
- 117 Huang AC, Postow MA, Orlowski RJ. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017; 545: 60-65
- 118 Zhang Z, Langenbach M, Sagar S. et al. Efficacy of CTLA-4 checkpoint therapy is dependent on IL-21 signaling to mediate cytotoxic reprogramming of PD-1+CD8+ T cells. Nat Immunol 2025; 26: 92-104
- 119 Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol 2007; 19: 408-415
- 120 Zajac AJ, Blattman JN, Murali-Krishna K. et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 1998; 188: 2205-2213
- 121 Zheng L, Qin S, Si W. et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021; 374: abe6474
- 122 Sen DR, Kaminski J, Barnitz RA. et al. The epigenetic landscape of T cell exhaustion. Science 2016; 354: 1165-1169
- 123 Wherry EJ, Ha S-J, Kaech SM. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27: 670-684
- 124 Paley MA, Kroy DC, Odorizzi PM. et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012; 338: 1220-1225
- 125 Speiser DE, Utzschneider DT, Oberle SG. et al. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion?. Nat Rev Immunol 2014; 14: 768-774
- 126 Bengsch B, Ohtani T, Khan O. et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018; 48: 1029-1045.e5
- 127 Siddiqui I, Schaeuble K, Chennupati V. et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019; 50: 195-211.e10
- 128 Utzschneider DT, Gabriel SS, Chisanga D. et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol 2020; 21: 1256-1266
- 129 Im SJ, Hashimoto M, Gerner MY. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016; 537: 417-421
- 130 Miller BC, Sen DR, Al Abosy R. et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019; 20: 326-336
- 131 Utzschneider DT, Charmoy M, Chennupati V. et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016; 45: 415-427
- 132 Wu T, Ji Y, Moseman EA. et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 2016; 1: eaai8593
- 133 Cheng Y, Gunasegaran B, Singh HD. et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 2021; 54: 1825-1840.e7
- 134 Chew V, Lai L, Pan L. et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A 2017; 114: E5900-E5909
- 135 Kim H-D, Song G-W, Park S. et al. Association Between Expression Level of PD1 by Tumor-Infiltrating CD8+ T Cells and Features of Hepatocellular Carcinoma. Gastroenterology 2018; 155: 1936-1950.e17
- 136 Kim H-D, Park S, Jeong S. et al. 4–1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Hepatology 2020; 71: 955-971
- 137 Zheng C, Zheng L, Yoo J-K. et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017; 169: 1342-1356.e16
- 138 Barsch M, Salié H, Mesesan A. et al. T cells in the heterogeneous tumour immune microenvironment of hepatocellular carcinoma: Implications for immune checkpoint inhibitor therapy. Liver Cancer International 2023; 4: 58-72
- 139 Hung MH, Lee JS, Ma C. et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun 2021; 12: 1455
- 140 Liu F, LiuWeirenSaninDavid E.. et al. Heterogeneity of exhausted T cells in the tumor microenvironment is linked to patient survival following resection in hepatocellular carcinoma. OncoImmunology 2020; 9: 1746573
- 141 Ma J, Zheng B, Goswami S. et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Cancer 2019; 7: 331
- 142 Steiner C, Denlinger N, Huang X. et al. Stem-like CD8+ T cells in cancer. Front Immunol 2024; 15: 1426418
- 143 Jang EJ, Choi HJ, You YK. et al. Differential Infiltration of T-Cell Populations in Tumor and Liver Tissues Predicts Recurrence-Free Survival in Surgically Resected Hepatocellular Carcinoma. Cancers (Basel) 2025; 17: 1548
- 144 Park M-S, Jo H, Kim H. et al. Molecular landscape of tumor-associated tissue-resident memory T cells in tumor microenvironment of hepatocellular carcinoma. Cell Commun Signal 2025; 23: 80
- 145 Yan F, Zhu B, Shi K. et al. Prognostic and therapeutic potential of imbalance between PD-1+CD8 and ICOS+Treg cells in advanced HBV-HCC. Cancer Sci 2024; 115: 2553-2564
- 146 Dudek M, Pfister D, Donakonda S. et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021; 592: 444-449
- 147 Feola S, Chiaro J, Martins B. et al. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers (Basel) 2020; 12: 1660
- 148 Dong L-Q, Peng L-H, Ma L-J. et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J Hepatol 2020; 72: 896-908
- 149 Lu L, Jiang J, Zhan M. et al. Targeting Tumor-Associated Antigens in Hepatocellular Carcinoma for Immunotherapy: Past Pitfalls and Future Strategies. Hepatology 2021; 73: 821-832
- 150 Flecken T, Schmidt N, Hild S. et al. Immunodominance and functional alterations of tumor‐associated antigen‐specific CD8+ T‐cell responses in hepatocellular carcinoma. Hepatology 2014; 59: 1415-1426
- 151 Zhou G, Sprengers D, Boor PPC. et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017; 153: 1107-1119.e10
- 152 Tauber C, Schultheiss M, Luca RD. et al. Inefficient induction of circulating TAA-specific CD8+ T-cell responses in hepatocellular carcinoma. Oncotarget 2019; 10: 5194-5206
- 153 Puig-Saus C, Sennino B, Peng S. et al. Neoantigen-targeted CD8+ T cell responses with PD-1 blockade therapy. Nature 2023; 615: 697-704
- 154 Xie N, Shen G, Gao W. et al. Neoantigens: promising targets for cancer therapy. Sig Transduct Target Ther 2023; 8: 1-38
- 155 Petrizzo A, Tagliamonte M, Mauriello A. et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J Transl Med 2018; 16: 286
- 156 Aggeletopoulou I, Pantzios S, Triantos C. Personalized Immunity: Neoantigen-Based Vaccines Revolutionizing Hepatocellular Carcinoma Treatment. Cancers 2025; 17: 376
- 157 Liu T, Tan J, Wu M. et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39+CD8+ T cells. Gut 2021; 70: 1965-1977
- 158 Maravelia P, Yao H, Cai C. et al. Unlocking novel T cell-based immunotherapy for hepatocellular carcinoma through neoantigen-driven T cell receptor isolation. Gut 2025; 74: 1125-1136
- 159 Rojas LA, Sethna Z, Soares KC. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023; 618: 144-150
- 160 Yarchoan M, Gane EJ, Marron TU. et al. Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: a phase 1/2 trial. Nat Med 2024; 30: 1044-1053
- 161 Pu S, Liu T, Gao Y. et al. The role of tumor-associated endothelial cells in malignant progression and immune evasion of liver cancer. Int Immunopharmacol 2025; 161: 115013
- 162 Zhou P-Y, Zhou C, Gan W. et al. Single-cell and spatial architecture of primary liver cancer. Commun Biol 2023; 6: 1181
- 163 Xie Z, Huang J, Li Y. et al. Single-cell RNA sequencing revealed potential targets for immunotherapy studies in hepatocellular carcinoma. Sci Rep 2023; 13: 18799
- 164 Wang Y-H, Cheng T-Y, Chen T-Y. et al. Plasmalemmal Vesicle Associated Protein (PLVAP) as a therapeutic target for treatment of hepatocellular carcinoma. BMC Cancer 2014; 14: 815
- 165 Carambia A, Freund B, Schwinge D. et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J Hepatol 2014; 61: 594-599
- 166 Lu Y, Liu Y, Zuo X. et al. CXCL12+ tumor-associated endothelial cells promote immune resistance in hepatocellular carcinoma. J Hepatol 2025; 82: 634-648
- 167 Gui M, Huang S, Li S. et al. Integrative single-cell transcriptomic analyses reveal the cellular ontological and functional heterogeneities of primary and metastatic liver tumors. J Transl Med 2024; 22: 206
- 168 Sas Z, Cendrowicz E, Weinhäuser I. et al. Tumor Microenvironment of Hepatocellular Carcinoma: Challenges and Opportunities for New Treatment Options. Int J Mol Sci 2022; 23: 3778
- 169 Kane K, Edwards D, Chen J. The influence of endothelial metabolic reprogramming on the tumor microenvironment. Oncogene 2025; 44: 51-63
- 170 Hack SP, Zhu AX, Wang Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front Immunol 2020; 11: 598877
- 171 Zhu AX, Abbas AR, de Galarreta MR. et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med 2022; 1-13
- 172 Baglieri J, Brenner DA, Kisseleva T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. International Journal of Molecular Sciences 2019; 20: 1723
- 173 Zhang DY, Goossens N, Guo J. et al. A hepatic stellate cell gene expression signature associated with outcomes in hepatitis C cirrhosis and hepatocellular carcinoma after curative resection. Gut 2016; 65: 1754-1764
- 174 Ying F, Chan MSM, Lee TKW. Cancer-Associated Fibroblasts in Hepatocellular Carcinoma and Cholangiocarcinoma. Cell Mol Gastroenterol Hepatol 2023; 15: 985-999
- 175 Chiavarina B, Ronca R, Otaka Y. et al. Fibroblast-derived prolargin is a tumor suppressor in hepatocellular carcinoma. Oncogene 2022; 41: 1410-1420
- 176 Roy AM, Iyer R, Chakraborty S. The extracellular matrix in hepatocellular carcinoma: Mechanisms and therapeutic vulnerability. Cell Rep Med 2023; 4: 101170
- 177 Lee B, Lee S-H, Shin K. Crosstalk between fibroblasts and T cells in immune networks. Front Immunol 2022; 13: 1103823
- 178 Lan X, Li W, Zhao K. et al. Revisiting the role of cancer-associated fibroblasts in tumor microenvironment. Front Immunol 2025; 16: 1582532
- 179 Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005; 8: 369-380
- 180 Zhu G-Q, Tang Z, Huang R. et al. CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov 2023; 9: 25
- 181 Bayo J, Real A, Fiore EJ. et al. IL-8, GRO and MCP-1 produced by hepatocellular carcinoma microenvironment determine the migratory capacity of human bone marrow-derived mesenchymal stromal cells without affecting tumor aggressiveness. Oncotarget 2017; 8: 80235-80248
- 182 Szoor A, Vaidya A, Velasquez MP. et al. T Cell-Activating Mesenchymal Stem Cells as a Biotherapeutic for HCC. Mol Ther Oncolytics 2017; 6: 69-79
- 183 Ohlsson LB, Varas L, Kjellman C. et al. Mesenchymal progenitor cell-mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol Pathol 2003; 75: 248-255
- 184 Gao W-X, Sun Y-Q, Shi J. et al. Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res Ther 2017; 8: 48
- 185 Selleri S, Bifsha P, Civini S. et al. Human mesenchymal stromal cell-secreted lactate induces M2-macrophage differentiation by metabolic reprogramming. Oncotarget 2016; 7: 30193-30210
- 186 Özdemir BC, Pentcheva-Hoang T, Carstens JL. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014; 25: 719-734
- 187 Haber PK, Puigvehí M, Castet F. et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021; 161: 879-898
- 188 Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S3-Leitlinie Diagnostik und Therapie des Hepatozellulären Karzinoms und biliärer Karzinome, Langversion 5.0, 2024, AWMF-Registernummer: 032–053OL. Accessed October 08, 2025 at: https://www.leitlinienprogramm-onkologie.de/leitlinien/hcc-und-biliaere-karzinome/
- 189 Sheng J, Zhang J, Wang L. et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut 2022; 71: 1176-1191
- 190 Shu DH, Ho WJ, Kagohara LT. et al. Immunotherapy response induces divergent tertiary lymphoid structure morphologies in hepatocellular carcinoma. Nat Immunol 2024; 25: 2110-2123
- 191 Salié H, Wischer L, D’Alessio A. et al. Spatial single-cell profiling and neighbourhood analysis reveal the determinants of immune architecture connected to checkpoint inhibitor therapy outcome in hepatocellular carcinoma. Gut 2025; 74: 451-466
Correspondence
Publication History
Received: 15 August 2025
Accepted after revision: 03 November 2025
Article published online:
26 January 2026
© 2026. Thieme. All rights reserved.
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 Zentrum für Krebsregisterdaten RKI. Krebs in Deutschland, p 44–47. 2023 Accessed August 10, 2025 at: https://www.krebsdaten.de/Krebs/DE/Content/Publikationen/Krebs_in_Deutschland/krebs_in_deutschland_2023.pdf?__blob=publicationFile
- 2 Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 1999; 19: 329-338
- 3 Reig M, Forner A, Rimola J. et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J Hepatol 2022; 76: 681-693
- 4 Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378-390
- 5 Cheng A-L, Kang Y-K, Chen Z. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. The Lancet Oncology 2009; 10
- 6 Kudo M, Finn RS, Qin S. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 2018; 391: 1163-1173
- 7 El-Khoueiry AB, Sangro B, Yau T. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. The Lancet 2017; 389: 2492-2502
- 8 Yau T, Park JW, Finn RS. et al. LBA38_PR – CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Annals of Oncology 2019; 30: v874-v875
- 9 Zhu AX, Finn RS, Edeline J. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. The Lancet Oncology 2018; 19: 940-952
- 10 Finn RS, Ryoo B-Y, Merle P. et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2020. Accessed August 03, 2020 at: https://pubmed.ncbi.nlm.nih.gov/31790344/
- 11 Finn RS, Qin S, Ikeda M. et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. The New England journal of medicine 2020. Accessed August 03, 2020 at: https://pubmed.ncbi.nlm.nih.gov/32402160/
- 12 Abou-Alfa GK, Lau G, Kudo M. et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evidence 2022; 1: EVIDoa2100070
- 13 Yau T, Galle PR, Decaens T. et al. Nivolumab plus ipilimumab versus lenvatinib or sorafenib as first-line treatment for unresectable hepatocellular carcinoma (CheckMate 9DW): an open-label, randomised, phase 3 trial. The Lancet 2025; 405: 1851-1864
- 14 Qin S, Kudo M, Meyer T. et al. Tislelizumab vs Sorafenib as First-Line Treatment for Unresectable Hepatocellular Carcinoma: A Phase 3 Randomized Clinical Trial. JAMA oncology 2023; 9
- 15 Kelley RK, Rimassa L, Cheng A-L. et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology 2022; 23
- 16 Llovet JM, Kudo M, Merle P. et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind, phase 3 trial. The Lancet Oncology 2023; 24
- 17 Qin S, Chan SL, Gu S. et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international phase 3 study. Lancet (London, England) 2023; 402
- 18 Scheiner B, Kang B, Balcar L. et al. Outcome and management of patients with hepatocellular carcinoma who achieved a complete response to immunotherapy-based systemic therapy. Hepatology 2025; 81: 1714-1727
- 19 Horst AK, Neumann K, Diehl L. et al. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol Immunol 2016; 13: 277-292
- 20 Zheng M, Tian Z. Liver-Mediated Adaptive Immune Tolerance. Front Immunol 2019; 10
- 21 Rennert C, Lang-Meli J, Gromak M. et al. Perspectives for novel therapeutic concepts in hepatocellular carcinoma targeting the stromal and innate immune microenvironment. Liver Cancer International 2023; 4: 42-57
- 22 Calderaro J, Ziol M, Paradis V. et al. Molecular and histological correlations in liver cancer. Journal of Hepatology 2019; 71: 616-630
- 23 Pfister D, Núñez NG, Pinyol R. et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021; 592: 450-456
- 24 Sharma A, Seow JJW, Dutertre C-A. et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020; 183: 377-394.e21
- 25 Lanzanò L, Coto Hernández I, Castello M. et al. Encoding and decoding spatio-temporal information for super-resolution microscopy. Nat Commun 2015; 6: 6701
- 26 Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011; 11: 762-774
- 27 Yang F, Wei Y, Han D. et al. Interaction with CD68 and Regulation of GAS6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res 2020; 80: 3892-3905
- 28 Yuan Y, Wu D, Li J. et al. Mechanisms of tumor-associated macrophages affecting the progression of hepatocellular carcinoma. Front Pharmacol 2023; 14
- 29 Liu P-S, Wang H, Li X. et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017; 18: 985-994
- 30 Tripathi C, Tewari BN, Kanchan RK. et al. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5: 5350-5368
- 31 Lu Y, Yang A, Quan C. et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun 2022; 13: 4594
- 32 Fan G, Xie T, Li L. et al. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol 2024; 8: 75
- 33 Tan J, Fan W, Liu T. et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J Hepatol 2023; 79: 126-140
- 34 Zhou L, Wang M, Guo H. et al. Integrated Analysis Highlights the Immunosuppressive Role of TREM2+ Macrophages in Hepatocellular Carcinoma. Front Immunol 2022; 13: 848367
- 35 Roderfeld M, Rath T, Lammert F. et al. Innovative immunohistochemistry identifies MMP-9 expressing macrophages at the invasive front of murine HCC. World J Hepatol 2010; 2: 175-179
- 36 Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 2011; 32: 19-25
- 37 Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res 2017; 5: 3-8
- 38 Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017; 8: 3649-3665
- 39 Mandruzzato S, Brandau S, Britten CM. et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother 2016; 65: 161-169
- 40 Bronte V, Brandau S, Chen S-H. et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7: 12150
- 41 Hoechst B, Ormandy LA, Ballmaier M. et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008; 135: 234-243
- 42 Arihara F, Mizukoshi E, Kitahara M. et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother 2013; 62: 1421-1430
- 43 Zhang X, Fu X, Li T. et al. The prognostic value of myeloid derived suppressor cell level in hepatocellular carcinoma: A systematic review and meta-analysis. PLoS One 2019; 14: e0225327
- 44 Ma C, Zhang Q, Greten TF. MDSCs in liver cancer: A critical tumor-promoting player and a potential therapeutic target. Cell Immunol 2021; 361: 104295
- 45 Chiu DK-C, Xu IM-J, Lai RK-H. et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016; 64: 797-813
- 46 Kasic T, Colombo P, Soldani C. et al. Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol 2011; 41: 1843-1849
- 47 Kusmartsev S, Nefedova Y, Yoder D. et al. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol 2004; 172: 989-999
- 48 Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol 2018; 9: 2499
- 49 Hoechst B, Voigtlaender T, Ormandy L. et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009; 50: 799-807
- 50 Lacotte S, Slits F, Orci LA. et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology 2016; 5: e1234565
- 51 Tomiyama T, Itoh S, Iseda N. et al. Myeloid‑derived suppressor cell infiltration is associated with a poor prognosis in patients with hepatocellular carcinoma. Oncology Letters 2022; 23: 1-9
- 52 Del Prete A, Salvi V, Soriani A. et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol 2023; 20: 432-447
- 53 Ormandy LA, Färber A, Cantz T. et al. Direct ex vivo analysis of dendritic cells in patients with hepatocellular carcinoma. World J Gastroenterol 2006; 12: 3275-3282
- 54 Saito Y, Komori S, Kotani T. et al. The Role of Type-2 Conventional Dendritic Cells in the Regulation of Tumor Immunity. Cancers (Basel) 2022; 14: 1976
- 55 Li W, Chen G, Peng H. et al. Research Progress on Dendritic Cells in Hepatocellular Carcinoma Immune Microenvironments. Biomolecules 2024; 14: 1161
- 56 Suthen S, Lim CJ, Nguyen PHD. et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology 2022;
- 57 Pang L, Yeung OWH, Ng KTP. et al. Postoperative Plasmacytoid Dendritic Cells Secrete IFNα to Promote Recruitment of Myeloid-Derived Suppressor Cells and Drive Hepatocellular Carcinoma Recurrence. Cancer Res 2022; 82: 4206-4218
- 58 Shi J-Y, Gao Q, Wang Z-C. et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin Cancer Res 2013; 19: 5994-6005
- 59 Brunner SM, Itzel T, Rubner C. et al. Tumor-infiltrating B cells producing antitumor active immunoglobulins in resected HCC prolong patient survival. Oncotarget 2017; 8: 71002-71011
- 60 Garnelo M, Tan A, Her Z. et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017; 66: 342-351
- 61 Trailin A, Červenková L, Ambrozkiewicz F. et al. T- and B-Cells in the Inner Invasive Margin of Hepatocellular Carcinoma after Resection Associate with Favorable Prognosis. Cancers (Basel) 2022; 14: 604
- 62 Faggioli F, Palagano E, Di Tommaso L. et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018; 67: 1970-1985
- 63 Wei Y, Lao X-M, Xiao X. et al. Plasma Cell Polarization to the Immunoglobulin G Phenotype in Hepatocellular Carcinomas Involves Epigenetic Alterations and Promotes Hepatoma Progression in Mice. Gastroenterology 2019; 156: 1890-1904.e16
- 64 Neo SY, Shuen TWH, Khare S. et al. Atypical memory B cells acquire Breg phenotypes in hepatocellular carcinoma. JCI Insight 2025; 10: e187025
- 65 Ouyang F-Z, Wu R-Q, Wei Y. et al. Dendritic cell-elicited B-cell activation fosters immune privilege via IL-10 signals in hepatocellular carcinoma. Nat Commun 2016; 7: 13453
- 66 Shao Y, Lo CM, Ling CC. et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett 2014; 355: 264-272
- 67 Xue H, Lin F, Tan H. et al. Overrepresentation of IL-10-Expressing B Cells Suppresses Cytotoxic CD4+ T Cell Activity in HBV-Induced Hepatocellular Carcinoma. PLoS One 2016; 11: e0154815
- 68 Xiao X, Lao X-M, Chen M-M. et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov 2016; 6: 546-559
- 69 Shalapour S, Lin X-J, Bastian IN. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017; 551: 340-345
- 70 Liu R-X, Wei Y, Zeng Q-H. et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology 2015; 62: 1779-1790
- 71 Sun Y, Wu P, Zhang Z. et al. Integrated multi-omics profiling to dissect the spatiotemporal evolution of metastatic hepatocellular carcinoma. Cancer Cell 2024; 42: 135-156.e17
- 72 Dusseaux M, Martin E, Serriari N. et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17–secreting T cells. Blood 2011; 117: 1250-1259
- 73 Heymann F, Tacke F. Immunology in the liver — from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016; 13: 88-110
- 74 Hudspeth K, Donadon M, Cimino M. et al. Human liver-resident CD56bright/CD16neg NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J Autoimmun 2016; 66: 40-50
- 75 Taketomi A, Shimada M, Shirabe K. et al. Natural killer cell activity in patients with hepatocellular carcinoma: a new prognostic indicator after hepatectomy. Cancer 1998; 83: 58-63
- 76 Xue J-S, Ding Z-N, Meng G-X. et al. The Prognostic Value of Natural Killer Cells and Their Receptors/Ligands in Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. Front Immunol 2022; 13: 872353
- 77 Jia G, He P, Dai T. et al. Spatial immune scoring system predicts hepatocellular carcinoma recurrence. Nature 2025; 640: 1031-1041
- 78 Molgora M, Bonavita E, Ponzetta A. et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017; 551: 110-114
- 79 Cai L, Zhang Z, Zhou L. et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol 2008; 129: 428-437
- 80 Sun H, Huang Q, Huang M. et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019; 70: 168-183
- 81 Yoshida Y, Yoshio S, Yamazoe T. et al. Phenotypic Characterization by Single-Cell Mass Cytometry of Human Intrahepatic and Peripheral NK Cells in Patients with Hepatocellular Carcinoma. Cells 2021; 10: 1495
- 82 Yu L, Liu X, Wang X. et al. TIGIT+ TIM-3+ NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus‑related hepatocellular carcinoma. Oncoimmunology 2021; 10: 1942673
- 83 Sun Y, Li T, Ding L. et al. Platelet-mediated circulating tumor cell evasion from natural killer cell killing through immune checkpoint CD155-TIGIT. Hepatology 2025; 81: 791-807
- 84 Rennert C, Tauber C, Fehrenbach P. et al. Adaptive Subsets Limit the Anti-Tumoral NK-Cell Activity in Hepatocellular Carcinoma. Cells 2021; 10: 1369
- 85 Wei H, Suo C, Gu X. et al. AKR1D1 suppresses liver cancer progression by promoting bile acid metabolism-mediated NK cell cytotoxicity. Cell Metab 2025; 37: 1103-1118.e7
- 86 Pesce S, Greppi M, Tabellini G. et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol 2017; 139: 335-346.e3
- 87 Treiner E, Duban L, Bahram S. et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003; 422: 164-169
- 88 Duan M, Goswami S, Shi J-Y. et al. Activated and Exhausted MAIT Cells Foster Disease Progression and Indicate Poor Outcome in Hepatocellular Carcinoma. Clinical Cancer Research 2019; 25: 3304-3316
- 89 Ruf B, Bruhns M, Babaei S. et al. Tumor-associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell 2023; 186: 3686-3705.e32
- 90 Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010; 28: 445-489
- 91 Hirano S, Iwashita Y, Sasaki A. et al. Increased mRNA expression of chemokines in hepatocellular carcinoma with tumor-infiltrating lymphocytes. Journal of Gastroenterology and Hepatology 2007; 22: 690-696
- 92 Budhu A, Forgues M, Ye Q-H. et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006; 10: 99-111
- 93 Ma C, Kesarwala AH, Eggert T. et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016; 531: 253-257
- 94 Zheng C, Snow BE, Elia AJ. et al. Tumor-specific cholinergic CD4+ T lymphocytes guide immunosurveillance of hepatocellular carcinoma. Nat Cancer 2023; 4: 1437-1454
- 95 Barsch M, Salié H, Schlaak AE. et al. T cell exhaustion and residency dynamics inform clinical outcomes in hepatocellular carcinoma. J Hepatol 2022;
- 96 Deenick EK, Ma CS. The regulation and role of T follicular helper cells in immunity. Immunology 2011; 134: 361-367
- 97 Magen A, Hamon P, Fiaschi N. et al. Intratumoral dendritic cell-CD4+ T helper cell niches enable CD8+ T cell differentiation following PD-1 blockade in hepatocellular carcinoma. Nat Med 2023; 29: 1389-1399
- 98 Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012; 72: 2162-2171
- 99 Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell 2023; 41: 450-465
- 100 Schmidt A, Oberle N, Krammer PH. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front Immunol 2012; 3
- 101 Huang Y, Liao H, Zhang Y. et al. Prognostic Value of Tumor-Infiltrating FoxP3+ T Cells in Gastrointestinal Cancers: A Meta Analysis. PLoS ONE 2014; 9: e94376
- 102 Lim CJ, Lee YH, Pan L. et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019; 68: 916-927
- 103 Tu J-F, Ding Y-H, Ying X-H. et al. Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep 2016; 6: 35056
- 104 Wang H, Zhang H, Wang Y. et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol 2021; 75: 1271-1283
- 105 Yang P, Li Q-J, Feng Y. et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012; 22: 291-303
- 106 Llovet JM, Bustamante J, Castells A. et al. Natural history of untreated nonsurgical hepatocellular carcinoma: rationale for the design and evaluation of therapeutic trials. Hepatology 1999; 29: 62-67
- 107 Schöniger-Hekele M, Müller C, Kutilek M. et al. Hepatocellular carcinoma in Central Europe: prognostic features and survival. Gut 2001; 48: 103-109
- 108 You M, Gao Y, Fu J. et al. Epigenetic regulation of HBV-specific tumor-infiltrating T cells in HBV-related HCC. Hepatology 2023; 78: 943-958
- 109 Giles JR, Globig A-M, Kaech SM. et al. CD8+ T cells in the cancer-immunity cycle. Immunity 2023; 56: 2231-2253
- 110 Barry M, Bleackley RC. Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2002; 2: 401-409
- 111 Koh C-H, Lee S, Kwak M. et al. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp Mol Med 2023; 55: 2287-2299
- 112 Gabrielson A, Wu Y, Wang H. et al. Intratumoral CD3 and CD8 T-cell Densities Associated with Relapse-Free Survival in HCC. Cancer Immunol Res 2016; 4: 419-430
- 113 Gao Q, Qiu S-J, Fan J. et al. Intratumoral Balance of Regulatory and Cytotoxic T Cells Is Associated With Prognosis of Hepatocellular Carcinoma After Resection. JCO 2007; 25: 2586-2593
- 114 Hofmann M, Tauber C, Hensel N. et al. CD8+ T Cell Responses during HCV Infection and HCC. J Clin Med 2021; 10: 991
- 115 McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 2019; 37: 457-495
- 116 Bengsch B, Johnson AL, Kurachi M. et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016; 45: 358-373
- 117 Huang AC, Postow MA, Orlowski RJ. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017; 545: 60-65
- 118 Zhang Z, Langenbach M, Sagar S. et al. Efficacy of CTLA-4 checkpoint therapy is dependent on IL-21 signaling to mediate cytotoxic reprogramming of PD-1+CD8+ T cells. Nat Immunol 2025; 26: 92-104
- 119 Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol 2007; 19: 408-415
- 120 Zajac AJ, Blattman JN, Murali-Krishna K. et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 1998; 188: 2205-2213
- 121 Zheng L, Qin S, Si W. et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021; 374: abe6474
- 122 Sen DR, Kaminski J, Barnitz RA. et al. The epigenetic landscape of T cell exhaustion. Science 2016; 354: 1165-1169
- 123 Wherry EJ, Ha S-J, Kaech SM. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27: 670-684
- 124 Paley MA, Kroy DC, Odorizzi PM. et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012; 338: 1220-1225
- 125 Speiser DE, Utzschneider DT, Oberle SG. et al. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion?. Nat Rev Immunol 2014; 14: 768-774
- 126 Bengsch B, Ohtani T, Khan O. et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 2018; 48: 1029-1045.e5
- 127 Siddiqui I, Schaeuble K, Chennupati V. et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019; 50: 195-211.e10
- 128 Utzschneider DT, Gabriel SS, Chisanga D. et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol 2020; 21: 1256-1266
- 129 Im SJ, Hashimoto M, Gerner MY. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016; 537: 417-421
- 130 Miller BC, Sen DR, Al Abosy R. et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019; 20: 326-336
- 131 Utzschneider DT, Charmoy M, Chennupati V. et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016; 45: 415-427
- 132 Wu T, Ji Y, Moseman EA. et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 2016; 1: eaai8593
- 133 Cheng Y, Gunasegaran B, Singh HD. et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 2021; 54: 1825-1840.e7
- 134 Chew V, Lai L, Pan L. et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A 2017; 114: E5900-E5909
- 135 Kim H-D, Song G-W, Park S. et al. Association Between Expression Level of PD1 by Tumor-Infiltrating CD8+ T Cells and Features of Hepatocellular Carcinoma. Gastroenterology 2018; 155: 1936-1950.e17
- 136 Kim H-D, Park S, Jeong S. et al. 4–1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Hepatology 2020; 71: 955-971
- 137 Zheng C, Zheng L, Yoo J-K. et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017; 169: 1342-1356.e16
- 138 Barsch M, Salié H, Mesesan A. et al. T cells in the heterogeneous tumour immune microenvironment of hepatocellular carcinoma: Implications for immune checkpoint inhibitor therapy. Liver Cancer International 2023; 4: 58-72
- 139 Hung MH, Lee JS, Ma C. et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun 2021; 12: 1455
- 140 Liu F, LiuWeirenSaninDavid E.. et al. Heterogeneity of exhausted T cells in the tumor microenvironment is linked to patient survival following resection in hepatocellular carcinoma. OncoImmunology 2020; 9: 1746573
- 141 Ma J, Zheng B, Goswami S. et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Cancer 2019; 7: 331
- 142 Steiner C, Denlinger N, Huang X. et al. Stem-like CD8+ T cells in cancer. Front Immunol 2024; 15: 1426418
- 143 Jang EJ, Choi HJ, You YK. et al. Differential Infiltration of T-Cell Populations in Tumor and Liver Tissues Predicts Recurrence-Free Survival in Surgically Resected Hepatocellular Carcinoma. Cancers (Basel) 2025; 17: 1548
- 144 Park M-S, Jo H, Kim H. et al. Molecular landscape of tumor-associated tissue-resident memory T cells in tumor microenvironment of hepatocellular carcinoma. Cell Commun Signal 2025; 23: 80
- 145 Yan F, Zhu B, Shi K. et al. Prognostic and therapeutic potential of imbalance between PD-1+CD8 and ICOS+Treg cells in advanced HBV-HCC. Cancer Sci 2024; 115: 2553-2564
- 146 Dudek M, Pfister D, Donakonda S. et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021; 592: 444-449
- 147 Feola S, Chiaro J, Martins B. et al. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers (Basel) 2020; 12: 1660
- 148 Dong L-Q, Peng L-H, Ma L-J. et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J Hepatol 2020; 72: 896-908
- 149 Lu L, Jiang J, Zhan M. et al. Targeting Tumor-Associated Antigens in Hepatocellular Carcinoma for Immunotherapy: Past Pitfalls and Future Strategies. Hepatology 2021; 73: 821-832
- 150 Flecken T, Schmidt N, Hild S. et al. Immunodominance and functional alterations of tumor‐associated antigen‐specific CD8+ T‐cell responses in hepatocellular carcinoma. Hepatology 2014; 59: 1415-1426
- 151 Zhou G, Sprengers D, Boor PPC. et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017; 153: 1107-1119.e10
- 152 Tauber C, Schultheiss M, Luca RD. et al. Inefficient induction of circulating TAA-specific CD8+ T-cell responses in hepatocellular carcinoma. Oncotarget 2019; 10: 5194-5206
- 153 Puig-Saus C, Sennino B, Peng S. et al. Neoantigen-targeted CD8+ T cell responses with PD-1 blockade therapy. Nature 2023; 615: 697-704
- 154 Xie N, Shen G, Gao W. et al. Neoantigens: promising targets for cancer therapy. Sig Transduct Target Ther 2023; 8: 1-38
- 155 Petrizzo A, Tagliamonte M, Mauriello A. et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J Transl Med 2018; 16: 286
- 156 Aggeletopoulou I, Pantzios S, Triantos C. Personalized Immunity: Neoantigen-Based Vaccines Revolutionizing Hepatocellular Carcinoma Treatment. Cancers 2025; 17: 376
- 157 Liu T, Tan J, Wu M. et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39+CD8+ T cells. Gut 2021; 70: 1965-1977
- 158 Maravelia P, Yao H, Cai C. et al. Unlocking novel T cell-based immunotherapy for hepatocellular carcinoma through neoantigen-driven T cell receptor isolation. Gut 2025; 74: 1125-1136
- 159 Rojas LA, Sethna Z, Soares KC. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023; 618: 144-150
- 160 Yarchoan M, Gane EJ, Marron TU. et al. Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: a phase 1/2 trial. Nat Med 2024; 30: 1044-1053
- 161 Pu S, Liu T, Gao Y. et al. The role of tumor-associated endothelial cells in malignant progression and immune evasion of liver cancer. Int Immunopharmacol 2025; 161: 115013
- 162 Zhou P-Y, Zhou C, Gan W. et al. Single-cell and spatial architecture of primary liver cancer. Commun Biol 2023; 6: 1181
- 163 Xie Z, Huang J, Li Y. et al. Single-cell RNA sequencing revealed potential targets for immunotherapy studies in hepatocellular carcinoma. Sci Rep 2023; 13: 18799
- 164 Wang Y-H, Cheng T-Y, Chen T-Y. et al. Plasmalemmal Vesicle Associated Protein (PLVAP) as a therapeutic target for treatment of hepatocellular carcinoma. BMC Cancer 2014; 14: 815
- 165 Carambia A, Freund B, Schwinge D. et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J Hepatol 2014; 61: 594-599
- 166 Lu Y, Liu Y, Zuo X. et al. CXCL12+ tumor-associated endothelial cells promote immune resistance in hepatocellular carcinoma. J Hepatol 2025; 82: 634-648
- 167 Gui M, Huang S, Li S. et al. Integrative single-cell transcriptomic analyses reveal the cellular ontological and functional heterogeneities of primary and metastatic liver tumors. J Transl Med 2024; 22: 206
- 168 Sas Z, Cendrowicz E, Weinhäuser I. et al. Tumor Microenvironment of Hepatocellular Carcinoma: Challenges and Opportunities for New Treatment Options. Int J Mol Sci 2022; 23: 3778
- 169 Kane K, Edwards D, Chen J. The influence of endothelial metabolic reprogramming on the tumor microenvironment. Oncogene 2025; 44: 51-63
- 170 Hack SP, Zhu AX, Wang Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front Immunol 2020; 11: 598877
- 171 Zhu AX, Abbas AR, de Galarreta MR. et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med 2022; 1-13
- 172 Baglieri J, Brenner DA, Kisseleva T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. International Journal of Molecular Sciences 2019; 20: 1723
- 173 Zhang DY, Goossens N, Guo J. et al. A hepatic stellate cell gene expression signature associated with outcomes in hepatitis C cirrhosis and hepatocellular carcinoma after curative resection. Gut 2016; 65: 1754-1764
- 174 Ying F, Chan MSM, Lee TKW. Cancer-Associated Fibroblasts in Hepatocellular Carcinoma and Cholangiocarcinoma. Cell Mol Gastroenterol Hepatol 2023; 15: 985-999
- 175 Chiavarina B, Ronca R, Otaka Y. et al. Fibroblast-derived prolargin is a tumor suppressor in hepatocellular carcinoma. Oncogene 2022; 41: 1410-1420
- 176 Roy AM, Iyer R, Chakraborty S. The extracellular matrix in hepatocellular carcinoma: Mechanisms and therapeutic vulnerability. Cell Rep Med 2023; 4: 101170
- 177 Lee B, Lee S-H, Shin K. Crosstalk between fibroblasts and T cells in immune networks. Front Immunol 2022; 13: 1103823
- 178 Lan X, Li W, Zhao K. et al. Revisiting the role of cancer-associated fibroblasts in tumor microenvironment. Front Immunol 2025; 16: 1582532
- 179 Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005; 8: 369-380
- 180 Zhu G-Q, Tang Z, Huang R. et al. CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov 2023; 9: 25
- 181 Bayo J, Real A, Fiore EJ. et al. IL-8, GRO and MCP-1 produced by hepatocellular carcinoma microenvironment determine the migratory capacity of human bone marrow-derived mesenchymal stromal cells without affecting tumor aggressiveness. Oncotarget 2017; 8: 80235-80248
- 182 Szoor A, Vaidya A, Velasquez MP. et al. T Cell-Activating Mesenchymal Stem Cells as a Biotherapeutic for HCC. Mol Ther Oncolytics 2017; 6: 69-79
- 183 Ohlsson LB, Varas L, Kjellman C. et al. Mesenchymal progenitor cell-mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol Pathol 2003; 75: 248-255
- 184 Gao W-X, Sun Y-Q, Shi J. et al. Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res Ther 2017; 8: 48
- 185 Selleri S, Bifsha P, Civini S. et al. Human mesenchymal stromal cell-secreted lactate induces M2-macrophage differentiation by metabolic reprogramming. Oncotarget 2016; 7: 30193-30210
- 186 Özdemir BC, Pentcheva-Hoang T, Carstens JL. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014; 25: 719-734
- 187 Haber PK, Puigvehí M, Castet F. et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021; 161: 879-898
- 188 Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S3-Leitlinie Diagnostik und Therapie des Hepatozellulären Karzinoms und biliärer Karzinome, Langversion 5.0, 2024, AWMF-Registernummer: 032–053OL. Accessed October 08, 2025 at: https://www.leitlinienprogramm-onkologie.de/leitlinien/hcc-und-biliaere-karzinome/
- 189 Sheng J, Zhang J, Wang L. et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut 2022; 71: 1176-1191
- 190 Shu DH, Ho WJ, Kagohara LT. et al. Immunotherapy response induces divergent tertiary lymphoid structure morphologies in hepatocellular carcinoma. Nat Immunol 2024; 25: 2110-2123
- 191 Salié H, Wischer L, D’Alessio A. et al. Spatial single-cell profiling and neighbourhood analysis reveal the determinants of immune architecture connected to checkpoint inhibitor therapy outcome in hepatocellular carcinoma. Gut 2025; 74: 451-466