

Subscribe to RSS
DOI: 10.1055/s-0043-103533
Das Dandy-Walker-Syndrom
Authors

Das Dandy-Walker-Syndrom zählt zu den häufigsten angeborenen zerebellären Missbildungen. Hinter dieser Bezeichnung verbirgt sich aber eine Reihe verschiedener Krankheitsbilder mit ganz unterschiedlicher Pathologie, Therapie und Prognose. Der folgende Artikel bringt Licht in die verwirrende Terminologie, erleichtert die Diagnosestellung und hilft bei der Wahl der adäquaten Therapie entsprechend der zugrunde liegenden Malformation.
Abkürzungen
Einleitung
Das Dandy-Walker-Syndrom ist eine der häufigsten angeborenen zerebellären Missbildungen und manifestiert sich typischerweise mit einem Hydrozephalus. Die Terminologie ist oft verwirrend und wird nicht selten falsch verwendet. Aufgrund der unterschiedlichen Variabilität kann die Unterscheidung von anderen Zysten im Bereich der hinteren Schädelgrube erschwert sein. So können Diagnosen und Prognosen fehlinterpretiert werden. Die Ursachen einschließlich der Pathophysiologie sind weitgehend unbekannt. Zudem besteht Uneinigkeit hinsichtlich des idealen therapeutischen Managements.
Zur klassischen Definition der Dandy-Walker-Malformation gehören folgende Befunde:
-
Hypoplasie des Kleinhirnwurms (Vermis)
-
Vergrößerung der hinteren Schädelgrube
-
Aufwärtsabweichung des Torkulars und des Sinus transversus
-
zystische Erweiterung des IV. Ventrikels
-
häufig Hydrozephalus
Die Dandy-Walker-Malformation wurde erstmals 1914 von Walter Dandy und Kenneth Blackfan bei einem 13 Monate alten Säugling mit fehlenden Foramina Luschkae und Magendii beschrieben [1]. Sie vermuteten, dass ein Verschluss dieser Öffnungen zu einer Zystenbildung im IV. Ventrikel führte, mit konsekutiver Erweiterung der hinteren Schädelgrube und Entwicklung eines Hydrozephalus. Im Jahr 1942 beschrieben Taggart und Walker 3 ähnliche Fälle und postulierten als Ursache für die Vermishypoplasie den erhöhten Zystendruck im IV. Ventrikel [2]. In einer genaueren Studie an 6 Fällen folgerte Benda 1954, dass nicht die Atresie der Foramina, sondern die Fehlbildung des Vermis die primäre Anomalie darstelle [3].
Aktuell sind die genauen zugrunde liegenden Ursachen des Dandy-Walker-Syndroms nicht bekannt. Diskutiert wird zurzeit eine Fehlentwicklung des Rautendachs, des Kleinhirns und/oder benachbarter Strukturen. Für diese Hypothese spricht auch, dass das Dandy-Walker-Syndrom mit anderen Missbildungen des zentralen Nervensystems assoziiert sein kann. Beispiele dafür sind etwa eine Agenesie des Corpus callosum, eine Holoprosenzephalie, Heterotopien, Neuralrohrdefekte usw. oder Syndrome mit Beteiligung weiterer Organsysteme (s. Hintergrundboxen).
Mit dem zentralen Nervensystem assoziierte Fehlbildungen beim Dandy-Walker-Syndrom (nach [4]):
-
Corpus-callosum-Agenesie
-
Agyrie
-
Aquäduktstenose
-
Arhinozephalie
-
Hirnstammfehlbildungen
-
Cavum septi pellucidi
-
Chiari-I-Malformation
-
Plexus-choroideus-Zyste
-
Enzephalozele
-
fusionierte Massa intermedia
-
Hamartome
-
Hemimegalenzephalie
-
Heterotopien
-
Holoprosenzephalie
-
Hydrozephalus
-
interhemisphärische Zysten
-
Lipome (hintere Schädelgrube, spinal)
-
Makrozephalie
-
Makrogyrie
-
Malformation der Kleinhirnkerne
-
neurokutane Melanosis
-
Meningozele
-
Mikrozephalie
-
Myelomeningozele
-
Polymikrogyrie
-
Syringomyelie
-
zerebelläre Folienanomalien
Einige assoziierte Syndrome bei Dandy-Walker-Syndrom (nach [4]):
-
Chondrodystrophie
-
chronische hereditäre Polyneuropathie
-
Down-Syndrom
-
Goldstone-Syndrom
-
Klippel-Feil-Syndrom
-
Marden-Walker-Syndrom
-
Neurofibromatose
-
neurokutane Melanose
-
Opitz-C-Syndrom
-
Oralfazialdigitalsyndrom
-
partielle Trisomie 21
-
Sjögren-Larsson-Syndrom
-
Smith-Lemli-Opitz-Syndrom
-
Tetrasomie 9
-
Trisomie 9
-
Turner-Syndrom
-
Walker-Warburg-Syndrom
Bis zu 44 % der Patienten mit einer Dandy-Walker-Malformation versterben früh. Zwischen 30 und 50 % haben einen normalen Intelligenzquotienten, abhängig von weiteren Fehlbildungen. Epilepsien sind nicht selten bei ausgeprägten Fehlbildungen des Kleinhirnwurms vorhanden. Je weniger ausgeprägt diese Fehlbildungen sind, desto besser ist die Kognition und desto geringer sind die motorischen Defizite [5].
Die Prognose und das intellektuelle Outcome des Dandy-Walker-Syndroms hängen wesentlich von den assoziierten Malformationen, von der Ausprägung der Vermismissbildung und von der Behandlung des Hydrozephalus ab.
Terminologie
Zystische Fehlbildungen in der hinteren Schädelgrube werden zusammengefasst unter den Synonymen „Dandy-Walker-Syndrom“, „Dandy-Walker-Komplex“ oder „Dandy-Walker-Spektrum“.
Der Begriff „Dandy-Walker“ führt immer wieder zu Verwirrungen, da unter diesem Begriff unterschiedliche Krankheitsbilder subsumiert werden und je nach Pathologie die entsprechende Therapie und Prognose erheblich variieren können.
Deshalb ist es wichtig, initial die richtige Diagnose zu stellen, um das Krankheitsbild entsprechend erfolgreich behandeln zu können.
Zu den zystischen Fehlbildungen der hinteren Schädelgrube gehören folgende Formen [6]:
-
„klassische“ Dandy-Walker-Malformation
-
Dandy-Walker-Syndrom bzw. -Komplex
-
hypoplastischer Vermis mit Rotation, ehemals „Dandy-Walker-Variante“
-
persistierende Blakeʼs-Pouch-Zyste
-
Megacisterna magna
-
Arachnoidalzyste (von anderen Autoren dazu gezählt [7])
Epidemiologie und Genetik
Die Inzidenz der Dandy-Walker-Malformation beträgt ca. 1:30 000 Geburten mit leichter weiblicher Dominanz. Zwischen 1 und 4 % des infantilen Hydrozephalus sind assoziiert mit einer Dandy-Walker-Malformation, ca. 80 % werden innerhalb des ersten Lebensjahrs diagnostiziert. Die Prävalenz unter Geschwistern beträgt ca. 1 – 2 %, autosomal- oder X-chromosomal-rezessive Erbgänge sind beschrieben [8]. Grinberg u. Mitarb. konnten als Ursache eine Deletion des Chromosoms 3q2 mit heterozygotem Verlust der zerebellären Gene ZIC1 und ZIC4 nachweisen (Zinc Finger of the Cerebellum Protein Familiy 1 und 4) [9]. Das Chromosom 6p25.3 ist für die normale Differenzierung und Migration der Rautengrube sowie des Rhombenzephalons verantwortlich. Ein spezifischer Verlust des Gens FOXC1 (Forkhead Box 1) mit Lokalisation in diesem Chromosom kann auch mit zerebellären Malformationen wie Dandy-Walker-Malformation, zerebellärer Vermishypoplasie und Megacisterna magna in Zusammenhang stehen [10].
Einzelne Formen
Dandy-Walker-Malformation
Diese isolierte infratentorielle zystische Malformation der hinteren Schädelgrube ist definiert über die klassische Trias
-
zystische Erweiterung des IV. Ventrikels mit
-
konsekutiver Erweiterung der hinteren Schädelgrube und
-
Fehlbildung des Kleinhirnwurms ([Abb. 1]).
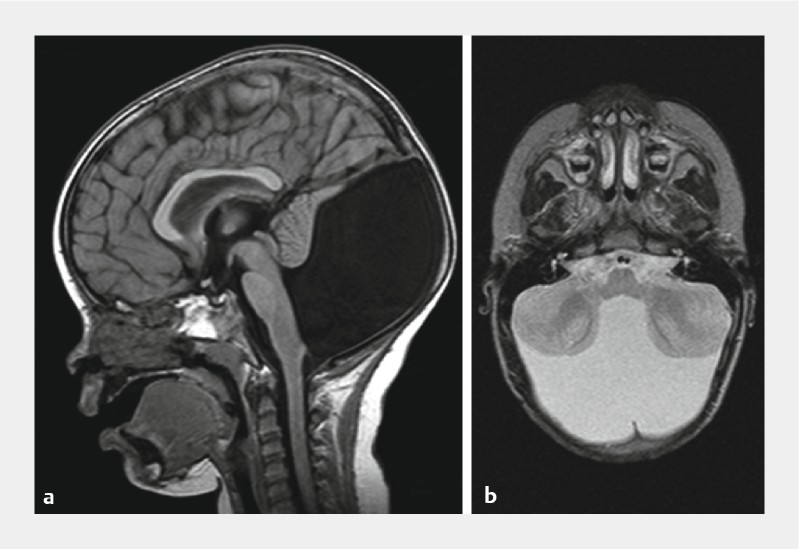
Letztere kann stark variieren und reicht von der Hypoplasie bis zur Agenesie. Zusätzlich ist eine Rotation nach anterior und superior vorhanden. Die Kleinhirnhemisphären sind meist hypoplastisch und können nach lateral abweichen. Der zerebelläre Kortex einschließlich der Kleinhirnkerne kann dysplastisch sein. Das gesamte Tentorium sowie die Sinus transversus und sigmoideus mitsamt dem Torkular sind nach oben verlagert. Entgegen der Theorie von Dandy, Walker u. Mitarb. sind die Foramina Luschkae und Magendii häufig offen. Der okzipitale Knochen ist oft ausgedünnt und der Klivus kann klein und konkav sein [8].
Dandy-Walker-Variante
Diese Form zeigt insgesamt eine mildere Ausprägung: Der IV. Ventrikel ist erweitert, nicht jedoch die hintere Schädelgrube. Die Vermishypoplasie ist weniger ausgeprägt und weniger häufig ist ein Hydrozephalus vorhanden ([Abb. 2]).

Blakeʼs-Pouch-Zyste
Bei der persistierenden Blakeʼs-Pouch-Zyste kommuniziert ein sog. offener IV. Ventrikel mit der Zyste, nicht aber mit dem Subarachnoidalraum ([Abb. 3]). Es handelt sich um eine inferiore Protrusion des IV. Ventrikels innerhalb der posterioren Membranacea (hintere Membran der ehemaligen Rautengrube). Bei normaler Entwicklung bildet sich zwischen der 7. und der 8. Gestationswoche in der Mitte vom IV. Ventrikel eine Öffnung kaudal zur inferioren Oberfläche des Kleinhirnwurms: das Foramen Magendii. Bildet sich diese Kommunikation nicht aus, so kommt es zu einer zystischen Erweiterung des IV. Ventrikels, ähnlich einer retrozerebellären Zyste. Dieser zystischen Erweiterung fehlt eine Verbindung zum Subarachnoidalraum. Die hintere Schädelgrube ist normal konfiguriert, Vermis und Kleinhirnhemisphären sind oft hypoplastisch, jedoch nicht rotiert. Das Tentorium kann partiell aufwärts verlagert, ein Hydrozephalus kann fakultativ vorhanden sein. Der Plexus choroideus des IV. Ventrikels ist häufig unterhalb der oberen Zystenwand zu sehen.

Megacisterna magna
Bei der Megacisterna magna (Synonym: Blakeʼs Continuum) wird die hintere Schädelgrube durch eine vergrößerte Cisterna magna zystisch erweitert ([Abb. 4]). Der Vermis ist intakt, die Kleinhirnhemisphären sind normal ausgebildet, ebenso wie der Hirnstamm. Typischerweise ist kein Hydrozephalus vorhanden. Es besteht eine freie Kommunikation über das Foramen Magendii mit dem IV. Ventrikel und über die Cisterna magna mit dem perimedullären Subarachnoidalraum. Das Tentorium kann partiell verlagert sein. Die Megacisterna magna ist relativ häufig und macht ca. die Hälfte aller Malformationen der hinteren Schädelgrube aus. In der Regel wird sie zufällig entdeckt, ist asymptomatisch und bedarf keiner weiteren operativen Therapie. Es gibt jedoch Untersuchungen, die bei Erwachsenen eine Beeinträchtigung höherer kognitiver und exekutiver Funktionen sowie der Sprache vermuten [11] [12].

Arachnoidalzyste
Die Arachnoidalzyste der hinteren Schädelgrube ist eine zystische, von Arachnoidalzellen umgebene Erweiterung mit und ohne Kontakt zum Subarachnoidalraum. Typische Lokalisationen sind retrozerebellär, supravermisch oder im Bereich des Kleinhirnbrückenwinkels. Arachnoidalzysten können angeboren oder erworben sein. Im letzteren Fall können sie durch arachnoidale Adhäsionen nach entzündlichen Prozessen im Rahmen von Traumata, Tumoren, Infektionen und/oder iatrogen entstanden sein [11]. Der IV. Ventrikel ist in der Regel normal entwickelt, kann aber von der Arachnoidalzyste komprimiert oder verlagert sein ([Abb. 5]). Oft handelt es sich um einen Zufallsbefund, der im Vergleich zu anderen Zystenlokalisationen aber häufiger klinisch symptomatisch sein kann. In der Regel sieht man eine scharf abgegrenzte, extraaxiale Flüssigkeitsansammlung von gleicher Dichte wie der Liquor. Arachnoidalzysten haben keine Verbindung zum IV. Ventrikel, das Kleinhirn kann je nach Zystengröße nach anterior verlagert sein.

Differenzialdiagnose
Bei der klassischen Dandy-Walker-Malformation müssen folgende Differenzialdiagnosen ausgeschlossen werden:
-
weitere (extra-)zerebrale Fehlbildungen
-
Konsanguinität der Eltern (bei der Anamnese erfragen!)
-
positive Familienanamnese
-
Chromosomendefekte (z. B. Trisomie 18, Triploidie, Trisomie 13 usw.)
Meist ist die Unterscheidung einer Blakeʼs-Pouch-Zyste von der Dandy-Walker-Malformation aufgrund des normal entwickelten Kleinhirns einfach.
Die Unterscheidung einer Arachnoidalzyste von der Dandy-Walker-Malformation ist anhand des normal entwickelten Vermis gut möglich. Diffusionsgewichtete MRT-Aufnahmen ermöglichen die Abgrenzung von Epidermoidzysten. Zu den Differenzialdiagnosen von Arachnoidalzysten gehört auch das Joubert-Syndrom mit ausgeprägter Vermishypoplasie und dem sog. Molar Tooth Sign (Backenzahnzeichen) mit vertiefter Fossa interpeduncularis und verdickten oberen Kleinhirnschenkeln ([Abb. 6]). Zusätzliche infra- und supratentorielle Veränderungen sind möglich. Das Joubert-Syndrom ist angeboren und genetisch sehr heterogen. Häufig liegen eine Muskelhypotonie, eine verzögerte allgemeine Entwicklung, kognitive Behinderungen und Ataxie vor. Eine Beteiligung von Retina, Leber und Nieren kann vorhanden sein und sollte in regelmäßigen Intervallen abgeklärt werden.

Zur Erleichterung der Differenzialdiagnose der einzelnen Entitäten des Dandy-Walker-Syndroms dient [Tab. 1].
Diagnostik
Klinik
Die meisten Patienten mit einer Dandy-Walker-Malformation werden während des ersten Lebensjahrs mit den typischen Zeichen des Hydrozephalus symptomatisch:
-
Makrozephalie bei Geburt bzw. deren rasche Entwicklung während des ersten Lebensjahrs
-
gespannte Fontanelle
-
Irritabilität und Erbrechen
-
klaffende Schädelnähte
-
deutliche Venenzeichnung
-
frontales Bossing (prominente Stirn)
-
Fehlen sog. Meilensteine in der Entwicklung
Ältere Kinder können Zeichen eines erhöhten intrakraniellen Druckes zeigen. Dazu gehören Stauungspapillen, Kopfschmerzen, rezidivierendes Erbrechen, Schwindel, Sehstörungen, Bewusstseinsstörungen, zerebrale Krampfanfälle, Parinaud-Syndrom usw. Es können aber auch Kleinhirnsymptome mit Ataxie und Nystagmus, Hirnnervenausfälle mit Doppelbildern, Schluckstörungen und Hörverlust auftreten. Auch Hypotonie, Spastik oder Sprachstörungen können beobachtet werden.
Bildgebende Verfahren
Im Ultraschall sind Malformationen der hinteren Schädelgrube schon ab der 14. Schwangerschaftswoche zu sehen, insbesondere mit dem 3D-Ultraschall. Bei Verdacht sollte ein fetales MRT angeschlossen werden. Pränatal kann eine Dandy-Walker-Malformation mit einem transvaginalen oder transabdominellen Ultraschall diagnostiziert werden. Eine signifikant schlechtere Prognose haben Feten, bei denen die Diagnose einer Dandy-Walker-Malformation bereits vor der 21. Schwangerschaftswoche gestellt werden kann [14]. Als Screening-Verfahren bei Säuglingen ist der Ultraschall speziell bei Makrozephalie ebenfalls geeignet zur Detektion von intrakraniellen Zysten und Hydrozephalus ([Abb. 7]). Eine Vermishypoplasie kann ggf. auch entdeckt werden, aber für eine detailliertere intrakranielle Darstellung ist dieses Verfahren nicht geeignet.

a Medianer Sagittalschnitt. Große, retrozerebellär lokalisierte Zyste, die mit dem dilatierten IV. Ventrikel kommuniziert. Hypoplasie und Dysplasie des Kleinhirnwurms. Gleichzeitig besteht eine Balkenagenesie, die zu einem dysplastischen, nach kranial verlagerten III. Ventrikel geführt hat. Fehlende Abbildung des Gyrus und Sulcus cinguli. Radiäre Anordnung der Sulki und Gyri um den dilatierten und dysplastischen III. Ventrikel als Zeichen der Balkenagenesie. b Hinterer Koronarschnitt durch die Kleinhirnhemisphären. Hypoplastische Kleinhirnhemisphären. Retrozerebellär lokalisierte Zyste, die mit dem IV. Ventrikel kommuniziert. Dilatierte Seitenventrikel. Nach kranial verlagerter dilatierter und dysplastischer III. Ventrikel aufgrund der gleichzeitig bestehenden Balkenagenesie. c Weit nach okzipital geneigter Koronarschnitt. Oberhalb der dysplastischen Kleinhirnhemisphären zeigt sich eine große, retrozerebellär lokalisierte Zyste, die in dieser Schnittebene die gesamte hintere Schädelgrube ausfüllt. Dilatierte Seitenventrikel, die aufgrund der gleichzeitig bestehenden Balkenagenesie parallel angeordnet sind. d Parasagittalschnitt durch den Seitenventrikel. Abnorm hoher Tentoriumsansatz. Dadurch wirkt der erweiterte Seitenventrikel nach ventral abgekippt. Große, retrozerebellär lokalisierte Zyste, hypoplastische Kleinhirnhemisphären, die am Unterrand des Tentorium cerebelli nachweisbar sind.
Mit der kranialen CT können in der Mittellinie lokalisierte Defekte des Kleinhirns bzw. des Vermis und andere Malformationen des Gehirns besser als im Ultraschall dargestellt werden. Aufgrund der Strahlenbelastung wird dieses Verfahren heutzutage jedoch nur bei besonderen Fragestellungen (z. B. knöcherne Verhältnisse) in Ausnahmesituationen angewendet.
Goldstandard ist die MRT. Damit können detailliert die neurale und vaskuläre Anatomie, Fehlbildungen wie Agyrien, Polymikrogyrien, eine Agenesie des Corpus callosum, okzipitale Enzephalozelen, Aquäduktstenosen, Spaltbildungen, zystische Malformationen, eine Syrinx usw. dargestellt werden. Mithilfe von CISS-Sequenzen (Constructive Interference in Steady State) können Zystenwände besser sichtbar gemacht werden. Die Kommunikation zwischen dem III. und dem IV. Ventrikel und der Umgebung kann via Cine-MRT und Liquorflussmessungen genau studiert werden. Pathologien lassen sich so sichtbar machen. Dies ist vor allem für die weitere Behandlungsplanung von entscheidender Bedeutung.
Mit der genauen Darstellung der Anatomie der hinteren Schädelgrube ist die MRT den übrigen radiologischen Verfahren überlegen. Nur mit ihr ist eine genaue Diagnose bzw. Differenzialdiagnose des Dandy-Walker-Syndroms zu stellen.
Besonderes Augenmerk sollte auf den Kleinhirnwurm (Fissurierung, Lobulierung), auf die Kommunikation und den Durchmesser zwischen dem retrozerebellären Raum und dem IV. Ventrikel sowie auf die Anatomie bzw. Lokalisation von Tentorium und Vermis gelegt werden.
Therapie
Nicht alle Dandy-Walker-Syndrome benötigen eine operative Therapie, so z. B. einige Formen der Dandy-Walker-Variante, die Blakeʼs-Pouch-Zyste oder die Megacisterna magna. Die Voraussetzung dafür ist, dass der Patient asymptomatisch ist. Eine neurochirurgische Intervention ist notwendig, wenn ein symptomatischer Hydrozephalus bzw. zerebelläre oder Hirnstammsymptome infolge des Druckes durch den zystisch erweiterten IV. Ventrikel vorhanden sind. Die Therapie bezieht sich also auf die Kontrolle des Hydrozephalus und der Zyste der hinteren Schädelgrube. Das Management ist jedoch alles andere als einfach und die Diskussionen dazu sind recht kontrovers. Bei den ersten Operationen wurde eine Membranexzision in der Zyste der hinteren Schädelgrube auf Höhe des Foramen magnum durchgeführt, um die Kommunikation zum Subarachnoidalraum wiederherzustellen [1] [2]. Aufgrund erhöhter Morbidität und Letalität wird dieses invasive Verfahren heutzutage jedoch nicht mehr angewendet.
Shunt-Implantation
Das derzeit am häufigsten eingesetzte Verfahren ist die Shunt-Implantation. Jedoch wird eine fortlaufende Diskussion über die optimale Positionierung des proximalen Katheters geführt:
-
im Seitenventrikel mit VP-Shunt (ventrikuloperitonealem Shunt),
-
im IV. Ventrikel mit sog. CP-Shunt (zystoperitonealem Shunt) oder
-
mit einer Kombination von beiden (VP/CP-Shunt).
Da häufig bei dem Dandy-Walker-Syndrom das Aquädukt offen ist, wird als First-Line-Therapie der VP-Shunt empfohlen. Dieser hat den Vorteil, dass er mit weniger Dislokationen und Migrationen als die Katheter im IV. Ventrikel behaftet ist. Andere Autoren bevorzugen jedoch den CP-Shunt, da mit seiner Hilfe ein physiologischer Abfluss über das Aquädukt geschaffen wird [16]. Klare Richtlinien dazu existieren nicht.
Risiken und Gefahren der Shunt-Implantation:
-
Überdrainagen mit Hämatomen in der hinteren Schädelgrube, eine transtentorielle Aufwärtsherniation, ein sog. Hirnstamm-Tethering mit diversen Ausfällen von kaudalen Hirnnerven (Schluckstörungen, Doppelbildern, Schielen, Nystagmus usw.) können bei beiden Shunt-Arten auftreten.
-
Bei der Einlage eines VP-Shunt über ein parietookzipitales Bohrloch sollte unbedingt die hohe Lage des Sinus transversus berücksichtigt werden, um Komplikationen mit Sinusverletzungen bis hin zu unstillbaren Blutungen zu verhindern.
-
Ferner besteht bei einem VP-Shunt mit Überdrainage eher die Gefahr eines Aquäduktverschlusses (speziell bei Verwendung von Ventilsystemen, die zu Überdrainagen neigen). In der Folge kann es zur Komplikation eines isolierten IV. Ventrikels mit Einklemmungsgefahr kommen.
-
Bei einem CP-Shunt hingegen besteht die Gefahr einer Katheterdislokation mit Verletzung des Bodens des IV. Ventrikels und Hirnnervenausfällen. Auch kann es zu Hämatomen im Bereich der hinteren Schädelgrube kommen oder zu einem Hirnstamm-Tethering.
Die Verwendung eines kombinierten VP/CP-Shunt empfehlen Raimondi u. Mitarb. [17] mit Verbindung eines supra- und eines infratentoriellen Katheters über einen Y-Konnektor. So lassen sich Druckunterschiede zwischen oberen und unteren Kompartimenten verhindern. Damit könnten Komplikationen wie eine transtentorielle Herniation, eine funktionelle Aquäduktstenose, ein supratentorieller Hydrozephalus und Hirnstammaffektionen vermieden werden.
Navigierte Kathetereinlage
Eine weitere Möglichkeit, wenn auch nicht ganz ungefährlich und abhängig von der Erfahrung des Operateurs, ist die navigierte Einlage eines Katheters in den Seitenventrikel durch das Foramen Monroi in die Zyste des IV. Ventrikels [18].
Drittventrikulostomie
Eine deutlich weniger invasive Therapieoption ist die endoskopische Drittventrikulostomie bei mit einem Dandy-Walker-Syndrom assoziiertem Hydrozephalus [19]. Dabei wird ein Loch in den Boden des III. Ventrikels angelegt, um eine Kommunikation zwischen den inneren und den äußeren Liquorräumen herzustellen. Voraussetzung dafür ist allerdings ein offenes Aquädukt. Zudem besteht eine veränderte Anatomie (Boden des III. Ventrikels nahezu vertikal und hoch liegende Basilarisspitze), die mitberücksichtigt werden muss.
Die Erfolgsrate der Drittventrikulostomie bei Kindern vor Beendigung des ersten Lebensjahrs ist aufgrund der schnellen Verschlussrate des Loches deutlich erniedrigt.
Bei Patienten mit bestehender Aquäduktstenose wird als Alternativoperation die Kombination aus endoskopischer Drittventrikulostomie und offener Zystenfensterung (s. unten) empfohlen [8].
Offene Zystenfensterung
In letzter Zeit ist auch die offene Zystenfensterung wieder in den Blickpunkt gerückt [20] [21] [22]. Dabei wird über eine Kraniotomie der hinteren Schädelgrube einschließlich einer Laminektomie des ersten Halswirbelkörpers eine weite Zystenfensterung bis in beide Kleinhirnbrückenwinkel und den zervikalen Subarachnoidalraum vorgenommen. Allerdings sollten die Kinder älter als 3 Jahre sein, da sie dann einen besser ausgebildeten zervikalen Subarachnoidalraum aufweisen. Größere Fallzahlen dazu existieren jedoch nicht.
Outcome/Prognose
In der Prä-CT-Ära betrug die Letalität der Dandy-Walker-Malformation (DWM) bis zu 50 % und verringerte sich nach Einführung der Liquorableitung mittels Shunt auf 10 – 20 % [23]. Die aktuelle Letalität wird mit 10 – 14 % angegeben [8]. Hauptgründe sind Infektionen, ein unkontrollierter Hydrozephalus und Shunt-Komplikationen. Eine schlechte Prognose haben Patienten mit zerebralen Krampfanfällen, signifikanten Hör- und Sehstörungen und anderen Anomalien. Das intellektuelle Outcome ist variabel und hängt von den assoziierten Fehlbildungen des zentralen Nervensystems ab. Bis zu 50 % der Betroffenen können sich normal entwickeln. Zahlreiche Diskussionen werden geführt, ob es eine Korrelation zwischen Vermisanomalie und Intelligenzquotient gibt [4] [24]. Obwohl einige Autoren den Einfluss des Hydrozephalus auf den Intelligenzquotienten verneinen [25], steigt die Komplikationsrate bei unbehandeltem und/oder nicht ausreichend therapiertem Hydrozephalus. Das ist prognostisch eher ungünstig.
Die Einführung von Shunts hat die Prognose des Dandy-Walker-Syndroms nachhaltig verbessert. Auch die verbesserte pränatale Diagnostik mit Ultraschall, Amniozentese sowie fetaler MRT konnte die Prognose und das Outcome verbessern und ermöglicht eine frühere Diagnose. Damit werden Therapieoptionen bereits frühzeitig verfügbar.
Fazit
Aufgrund der Variabilität der einzelnen Missbildungen des Dandy-Walker-Formenkreises sowie möglicher zusätzlicher anderer Hirnfehlbildungen ist die Diagnose einschließlich Differenzialdiagnose oft schwierig und verwirrend. Entsprechend sind auch die Prognosen und die therapeutischen Strategien unterschiedlich. Deshalb ist es wichtig, die anatomischen Unterschiede dieser Entitäten zu kennen, um die richtige Diagnose zu stellen und die weitere Therapie zu planen. Mit verbesserter pränataler Diagnostik mittels Ultraschall und Amniozentese sowie fetaler MRT kann heutzutage die Diagnose früh gestellt werden. Eine adäquate Therapie kann frühzeitig eingeleitet werden. Entscheidend für das weitere Outcome sind Kenntnisse über die Symptome und die erfolgreiche Behandlung des Hydrozephalus.
-
Zystische Fehlbildungen der hinteren Schädelgrube werden unter dem Begriff „Dandy-Walker“ zusammengefasst. Typische Manifestation ist der Hydrozephalus.
-
Zum Dandy-Walker-Formenkreis zählen die klassische Dandy-Walker-Malformation, die Dandy-Walker-Variante, die persistierende Blakeʼs-Pouch-Zyste, die Megacisterna magna und bei manchen Autoren die Arachnoidalzyste der hinteren Schädelgrube.
-
Die klassische Dandy-Walker-Malformation geht mit einer Erweiterung des IV. Ventrikels und der hinteren Schädelgrube sowie einer Vermisfehlbildung einher. Bei der Dandy-Walker-Variante hingegen ist die hintere Schädelgrube nicht erweitert und die Vermishypoplasie ist weniger ausgeprägt. Bei der Blakeʼs-Pouch-Zyste ist das Kleinhirn im Gegensatz zur Dandy-Walker-Malformation normal entwickelt. Ein Hydrozephalus kann vorhanden sein, während er bei der Megacisterna magna typischerweise fehlt. Eine Arachnoidalzyste in der hinteren Schädelgrube kann anhand des normal entwickelten Vermis gut von der Dandy-Walker-Malformation abgegrenzt werden.
-
Die Symptome einer Dandy-Walker-Malformation manifestieren sich typischerweise klinisch im Verlauf des ersten Lebensjahrs als Anzeichen eines Hydrozephalus. In der Bildgebung wird vor allem der Ultraschall eingesetzt, das kraniale CT wegen der Strahlenbelastung nur bei speziellen Fragestellungen. Goldstandard der Bildgebung ist die MRT.
-
Bei symptomatischen Patienten gibt es mehrere unterschiedlich invasive und komplikationsträchtige Therapieoptionen, so u. a. die Shunt-Implantation, die Drittventrikulostomie und die offene Zystenfensterung.
-
Die Prognose und das intellektuelle Outcome hängen wesentlich von den assoziierten Malformationen, von der Ausprägung der Vermismissbildung und von der möglichst frühzeitigen Behandlung des Hydrozephalus ab.
Erstveröffentlichung
Dieser Beitrag wurde erstveröffentlicht in: Neurochirurgie Scan 2016; 4: 129 – 139.
Über den Autor
Heidi Bächli

PD Dr. med.; Ausbildung zur Fachärztin für Neurochirurgie im Universitätsspital Basel sowie in der Klinik und Poliklinik für Neurochirurgie, Universität Würzburg. 2005–2012 Aufbau und Leitung der Pädiatrischen Neurochirurgie im Universitätskinderspital beider Basel (UKBB). Seit 2010 Leiterin der Pädiatrischen Neurochirurgie des Universitätsklinikums Heidelberg. Schwerpunkte: pädiatrische Neurochirurgie, Hydrozephalus, kraniale Missbildungen usw.
Interessenkonflikt
Die Autorin gibt an, dass kein Interessenkonflikt vorliegt.
-
Literatur
- 1 Dandy W, Blackfan K. Internal hydrocephalus: an experimental, clinical and pathological study. Am J Dis Child 1914; 406-482
- 2 Taggart TK, Waker AE. Congenital atresia of foramina of Luschka and Magendie. Arch Neurol Psychiatry 1942; 583-612
- 3 Benda CE. The Dandy-Walker syndrome or the so-called atresia of the foramen Magendie. J Neuropathol Exp Neurol 1954; 13: 14-29
- 4 Raimondi AJ, Sato K, Shimoji T. Dandy-Walker Syndrome. Basel: S. Karger; 1984: 21-45
- 5 Klein O, Pierre-Kahn A, Boddaert N. et al. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst 2003; 19: 484-489
- 6 Blaser SI. Dandy-Walker continuum. In: Osborn AG, Salzman KL, Barkovich AJ. , eds. Diagnostic imaging brain. 2nd. ed. Philadelphia: Amirsys; 2010: 26-29
- 7 Tortori-Donati P, Rossi A, Biancheri R. Brain malformations. In: Tortori-Donati P, Rossi A. , eds. Pediatric neuroradiology brain. Berlin: Springer; 2005: 138-155
- 8 Wilkinson CC, Winston KR. Congenital arachnoid cysts and the Dandy-Walker complex. In: Albright AL, Pollack IF, Adelson PD. , eds. Principles and practice of pediatric neurosurgery. Stuttgart: Thieme; 2007: 162-186
- 9 Grinberg I, Northrup H, Ardinger H. et al. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet 2004; 36: 1053-1055
- 10 Aldinger KA, Lehmann OJ, Hudgins L. et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet 2009; 41: 1037-1042
- 11 Correa GG, Amaral LF, Vedolin LM. Neuroimaging of Dandy-Walker malformation: new concepts. Top Magn Reson Imaging 2011; 22: 303-312
- 12 Bolduc ME, Limperopoulos C. Neurodevelopmental outcomes in children with cerebellar malformations: a systematic review. Dev Med Child Neurol 2009; 51: 256-267
- 13 Albright AL, Pollak IF, Adelson PD. Principles and practice of pediatric neurosurgery. New York: Thieme; 2015
- 14 Ulm B, Ulm MR, Deutinger J. et al. Dandy-Walker malformation: prenatal diagnosis in two consecutive pregnancies. AM J Perinatol 1999; 16: 61-63
- 15 Deeg KH, Hofmann V, Hoyer PF. Ultraschalldiagnostik in Pädiatrie und Kinderchirurgie. 4. Aufl. Stuttgart: Thieme; 2013
- 16 Asai A, Hoffmann H, Hendrick EB. et al. Dandy-Walker syndrome: experience at the Hospital for Sick Children, Toronto. Pediatr Neurosci 1989; 15: 66-73
- 17 Raimondi AJ, Samuelson G, Yarzagaray L. et al. Atresia of the foramina of Luschka and Magendie: the Dandy-Walker cyst. J Neurosurg 1969; 31: 202-216
- 18 Cedzich C, Lunkenheimer A, Baier G. et al. Ultrasound-guided puncture of a Dandy-Walker cyst via the lateral and III ventricles. Childs Nerv Syst 1999; 15: 472-476
- 19 Warf BC, Dewan M, Mugamba J. Management of Dandy-Walker complex-associated infant hydrocephalus by combined endoscopic third ventriculostomy and choroid plexus cauterization. J Neurosurg Pediatrics 2011; 8: 377-383
- 20 Liu JC, Ciacci JD, George TM. Brainstem tethering in Dandy-Walker syndrome: a complication of cystoperitoneal shunting. Case report. J Neurosurg 1995; 83: 1072-1074
- 21 Almeida GM, Matushita H, Mattosinho-Franca SMK. Dandy-Walker syndrome: posterior fossa craniectomy and cyst fenestration after several shunt revisions. Child Nerv Syst 1990; 6: 335-337
- 22 Villavicencio AT, Wellos JC, George TM. Avoiding complicated shunt systems by open fenestration of symptomatic fourth ventricular cysts associated with hydrocephalus. Pediatr Neurosurg 1998; 29: 314-319
- 23 Spennato P, Mirone G, Nastro A. et al. Hydrocephalus in Dandy-Walker malformation. Childs Nerv Syst 2011; 27: 1665-1681
- 24 Boddaert N, Klein O, Ferguson N. et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology 2003; 45: 320-324
- 25 Bindal AK, Storrs BB, McLone DG. Management of Dandy-Walker syndrome. Pediatr Neurosurg 1990; 16: 163-169
Korrespondenzadresse
-
Literatur
- 1 Dandy W, Blackfan K. Internal hydrocephalus: an experimental, clinical and pathological study. Am J Dis Child 1914; 406-482
- 2 Taggart TK, Waker AE. Congenital atresia of foramina of Luschka and Magendie. Arch Neurol Psychiatry 1942; 583-612
- 3 Benda CE. The Dandy-Walker syndrome or the so-called atresia of the foramen Magendie. J Neuropathol Exp Neurol 1954; 13: 14-29
- 4 Raimondi AJ, Sato K, Shimoji T. Dandy-Walker Syndrome. Basel: S. Karger; 1984: 21-45
- 5 Klein O, Pierre-Kahn A, Boddaert N. et al. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst 2003; 19: 484-489
- 6 Blaser SI. Dandy-Walker continuum. In: Osborn AG, Salzman KL, Barkovich AJ. , eds. Diagnostic imaging brain. 2nd. ed. Philadelphia: Amirsys; 2010: 26-29
- 7 Tortori-Donati P, Rossi A, Biancheri R. Brain malformations. In: Tortori-Donati P, Rossi A. , eds. Pediatric neuroradiology brain. Berlin: Springer; 2005: 138-155
- 8 Wilkinson CC, Winston KR. Congenital arachnoid cysts and the Dandy-Walker complex. In: Albright AL, Pollack IF, Adelson PD. , eds. Principles and practice of pediatric neurosurgery. Stuttgart: Thieme; 2007: 162-186
- 9 Grinberg I, Northrup H, Ardinger H. et al. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet 2004; 36: 1053-1055
- 10 Aldinger KA, Lehmann OJ, Hudgins L. et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet 2009; 41: 1037-1042
- 11 Correa GG, Amaral LF, Vedolin LM. Neuroimaging of Dandy-Walker malformation: new concepts. Top Magn Reson Imaging 2011; 22: 303-312
- 12 Bolduc ME, Limperopoulos C. Neurodevelopmental outcomes in children with cerebellar malformations: a systematic review. Dev Med Child Neurol 2009; 51: 256-267
- 13 Albright AL, Pollak IF, Adelson PD. Principles and practice of pediatric neurosurgery. New York: Thieme; 2015
- 14 Ulm B, Ulm MR, Deutinger J. et al. Dandy-Walker malformation: prenatal diagnosis in two consecutive pregnancies. AM J Perinatol 1999; 16: 61-63
- 15 Deeg KH, Hofmann V, Hoyer PF. Ultraschalldiagnostik in Pädiatrie und Kinderchirurgie. 4. Aufl. Stuttgart: Thieme; 2013
- 16 Asai A, Hoffmann H, Hendrick EB. et al. Dandy-Walker syndrome: experience at the Hospital for Sick Children, Toronto. Pediatr Neurosci 1989; 15: 66-73
- 17 Raimondi AJ, Samuelson G, Yarzagaray L. et al. Atresia of the foramina of Luschka and Magendie: the Dandy-Walker cyst. J Neurosurg 1969; 31: 202-216
- 18 Cedzich C, Lunkenheimer A, Baier G. et al. Ultrasound-guided puncture of a Dandy-Walker cyst via the lateral and III ventricles. Childs Nerv Syst 1999; 15: 472-476
- 19 Warf BC, Dewan M, Mugamba J. Management of Dandy-Walker complex-associated infant hydrocephalus by combined endoscopic third ventriculostomy and choroid plexus cauterization. J Neurosurg Pediatrics 2011; 8: 377-383
- 20 Liu JC, Ciacci JD, George TM. Brainstem tethering in Dandy-Walker syndrome: a complication of cystoperitoneal shunting. Case report. J Neurosurg 1995; 83: 1072-1074
- 21 Almeida GM, Matushita H, Mattosinho-Franca SMK. Dandy-Walker syndrome: posterior fossa craniectomy and cyst fenestration after several shunt revisions. Child Nerv Syst 1990; 6: 335-337
- 22 Villavicencio AT, Wellos JC, George TM. Avoiding complicated shunt systems by open fenestration of symptomatic fourth ventricular cysts associated with hydrocephalus. Pediatr Neurosurg 1998; 29: 314-319
- 23 Spennato P, Mirone G, Nastro A. et al. Hydrocephalus in Dandy-Walker malformation. Childs Nerv Syst 2011; 27: 1665-1681
- 24 Boddaert N, Klein O, Ferguson N. et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology 2003; 45: 320-324
- 25 Bindal AK, Storrs BB, McLone DG. Management of Dandy-Walker syndrome. Pediatr Neurosurg 1990; 16: 163-169
a Medianer Sagittalschnitt. Große, retrozerebellär lokalisierte Zyste, die mit dem dilatierten IV. Ventrikel kommuniziert. Hypoplasie und Dysplasie des Kleinhirnwurms. Gleichzeitig besteht eine Balkenagenesie, die zu einem dysplastischen, nach kranial verlagerten III. Ventrikel geführt hat. Fehlende Abbildung des Gyrus und Sulcus cinguli. Radiäre Anordnung der Sulki und Gyri um den dilatierten und dysplastischen III. Ventrikel als Zeichen der Balkenagenesie. b Hinterer Koronarschnitt durch die Kleinhirnhemisphären. Hypoplastische Kleinhirnhemisphären. Retrozerebellär lokalisierte Zyste, die mit dem IV. Ventrikel kommuniziert. Dilatierte Seitenventrikel. Nach kranial verlagerter dilatierter und dysplastischer III. Ventrikel aufgrund der gleichzeitig bestehenden Balkenagenesie. c Weit nach okzipital geneigter Koronarschnitt. Oberhalb der dysplastischen Kleinhirnhemisphären zeigt sich eine große, retrozerebellär lokalisierte Zyste, die in dieser Schnittebene die gesamte hintere Schädelgrube ausfüllt. Dilatierte Seitenventrikel, die aufgrund der gleichzeitig bestehenden Balkenagenesie parallel angeordnet sind. d Parasagittalschnitt durch den Seitenventrikel. Abnorm hoher Tentoriumsansatz. Dadurch wirkt der erweiterte Seitenventrikel nach ventral abgekippt. Große, retrozerebellär lokalisierte Zyste, hypoplastische Kleinhirnhemisphären, die am Unterrand des Tentorium cerebelli nachweisbar sind.