

RSS-Feed abonnieren
DOI: 10.1055/s-0045-1814368
The cerebellum in dystonia: key player or background support?
Autor*innen

Abstract
Since the 1960s, the pathophysiology of dystonia has been primarily attributed to dysfunction of the basal ganglia and their associated pathways. However, growing evidence from both basic and clinical research has highlighted the additional importance of the cerebellum, suggesting that dystonia arises from a motor-network dysfunction involving not only the basal ganglia, but also the cerebellum. Neuroimaging studies reinforce this concept, revealing structural and functional abnormalities in the cerebellum and its afferent pathways in patients with dystonia. Moreover, the dual involvement of the cerebellum and basal ganglia may help explain the frequent co-occurrence of dystonia in patients with ataxia and vice versa. The present review aims to integrate evidence from pathophysiology, clinical studies, genetics, and neuroimaging to underscore the crucial role of the cerebellum in the genesis of dystonia.
Keywords
Dystonia - Cerebellum - Purkinje Cells - Inferior Olivary Complex - Cholinergic Antagonists - Basal GangliaINTRODUCTION
The term dystonia derives a combination of the Latin prefix dis- and the Greek suffix tónos. The word tone originally carried musical connotations. It stems from the 13th-century Old French ton (of the voice), from the Latin tonus—meaning stretching, quality of sound, tone, or accent—which, in turn, also derives from the Greek suffix tónos, translated as stretching, tension, raising of the voice, or pitch.[1] According to the most recent consensus, dystonia is a movement disorder characterized by sustained or intermittent abnormal movements and/or postures. Dystonic movements and postures are typically patterned and repetitive, and they may appear tremulous or jerky. They are often triggered or exacerbated by voluntary actions, and they are frequently accompanied by overflow movements.[2]
Since the 1960s, the pathophysiology of dystonia has been primarily associated with dysfunction of the basal ganglia and their pathways.[1] However, growing evidence from both basic and clinical research[3] [4] has highlighted the additional importance of the cerebellum, suggesting that dystonia may result from a motor-network dysfunction involving not only the basal ganglia, but also the cerebellum.
The current narrative review aims to synthesize and organize the findings from various studies concerning the pathophysiology, genetics, clinical manifestations, neuroimaging, and treatment approaches that highlight the cerebellum's crucial role in the genesis of dystonia.
METHODS
The current narrative review included original research articles, such as observational, cohort, cross-sectional, and case–control studies, as well as case series, clinical cases, meta-analyses, and reviews that addressed the interplay among genetics, clinical findings, neurophysiology, neuroimaging, and the cerebellar pathophysiology of dystonia.
A three-step search strategy was employed to ensure comprehensive coverage of the literature. First, a preliminary search was conducted in major databases (PubMed, Embase, and the Cumulative Index to Nursing and Allied Health Literature [CINAHL]) using the term dystonia combined with ataxia and cerebellum. This stage aimed to identify index terms, Medical Subject Headings (MeSH), and keywords from titles and abstracts of the retrieved papers. Second, a full search was performed using all identified keywords and index terms across the selected databases. Third, the reference lists of all retrieved studies were manually reviewed to identify additional relevant publications not captured by the electronic searches. Two reviewers (CHFC and HAGT) independently screened all titles and abstracts, resolving discrepancies through discussion. In addition, the references of the selected studies were carefully examined to identify further relevant sources.
Complementary to this strategy, the authors reviewed published articles and books on the history of dystonia, covering the earliest investigations into its pathophysiology and its proposed relationship with the cerebellum. The objective was to develop a narrative that critically examined the evolution of this relationship over time and introduced subsequent topics in a logical and coherent sequence.
HISTORICAL ASPECTS
The term dystonia was first coined by Hermann Oppenheim (1858–1919) in his 1911 paper,[5] in which he described four unrelated children, all of Jewish origin, from Galicia (in Western Ukraine) and Russia. The standard features among these patients included twisted postures, intermittent sustained spasms, rapid and rhythmic movements, development of fixed postural deformities, and worsening during walking, all in the absence of weakness. Although Oppenheim initially considered hysteria or idiopathic bilateral athetosis as possible diagnoses, he ultimately recognized this as a new hereditary disorder and proposed the name dystonia musculorum deformans.[5]
As Oppenheim initially hypothesized with hysteria, dystonia often lingered under an ambiguous definition between a neurological and a psychiatric disorder. At times, it was even proposed that dystonia should not be considered a distinct movement disorder.[1] Jean-Martin Charcot (1825–1893), known as the “Father of Neurology,” showed a great interest in the study of hysteria and frequently presented patients with movement abnormalities that would today be classified as functional movement disorders, including functional dystonias.[6] In his 1908 doctoral thesis,[7] Marcus Walter Schwalbe (1883-1926) had already recognized dystonia as a condition distinct from previously-described movement disorders. He also identified its hereditary nature, which he observed in a Jewish family. Nevertheless, Schwalbe considered the disorder to be partly psychiatric, referring to it as a “tonic cramps syndrome with hysterical symptoms.”[7]
Oppenheim also considered the possibility of diagnosing athetosis.[5] The term athetosis, from the Greek meaning “without fixed position,” had been coined earlier, in 1871, by William Alexander Hammond (1828–1900), author of the first American neurology textbook. Although Hammond struggled to establish athetosis as a distinct clinicopathological entity—and despite his accurate prediction of striatal pathology in his initial case—many neurologists regarded athetosis as merely a form of post-hemiplegic chorea or as part of a chorea–dystonia continuum.[8] [9] Nevertheless, his pathological observations encouraged contemporary physicians and researchers to associate such movement disorders with irritation or damage to the basal ganglia and thalamus, rather than with pyramidal tract dysfunction.[9]
Despite this early understanding of the role of the basal ganglia in movement disorders—and the vigorous debate nearly 4 decades later on the very nosographic status of dystonia at the Tenth International Neurological Meeting in Paris, in 1929, with contributions from Ludo van Bogaert (1897–1989), Jules Froment (1878–1946), Gheorghe Marinescu (1863–1938), Jean Alexandre Barré (1880–1967), Henry Meige (1866–1940), and Jean Lhermitte (1877–1959)—neurological interest in dystonia subsequently waned for more than a decade. During this period, psychiatrists, strongly influenced by psychoanalytic thought, advanced Freudian explanations for the contorted and disfiguring postures observed in the focal and generalized forms of dystonia.[1] [9]
This situation began to change between the 1940s and 1970s, when clinicians such as Ernest Herz (1901–1966) and Wolfgang Zeman (1921–2001) revisited the phenomenology of dystonia, emphasizing its neurological basis. Their detailed clinical observations highlighted the organic nature of the disorder and paved the way for a renewed interest in its classification.[10] [11] During the same period, Derek Denny-Brown (1901–1981) published 3 classic monographs: Diseases of the Basal Ganglia and Subthalamic Nuclei (1946), The Basal Ganglia (1962), and The Cerebral Control of Movement (1966). These works provided, for the first time, an in-depth discussion of the anatomy and physiology of the basal ganglia in relation to several movement disorders. In his most influential volume, The Basal Ganglia and Their Relation to Disorders of Movement, he also examined classic diseases such as dystonias.[12] The true revival of interest, however, occurred in the 1970s, driven mainly by David Marsden (1938–1998), who redefined the phenomenology, emphasized the variability in clinical presentations, and reframed dystonia as a primary movement disorder caused by dysfunction of the basal ganglia.[13]
In 1886, William Gowers (1845–1915) reported that writer's cramp could sometimes be preceded by trauma or local disease.[14] According to the current understanding, following a peripheral injury, any alteration in normal anatomy and physiology may result in peripherally-induced movement disorders (PIMDs), which also include certain task-specific disorders arising after repetitive activities, often referred to as overuse syndromes. Some examples are musicians' dystonia, sports-related dystonia, and other movement disorders in performing artists, which often develop after years of highly-skilled, precise motor practice, or following a recent increase in training intensity.[15]
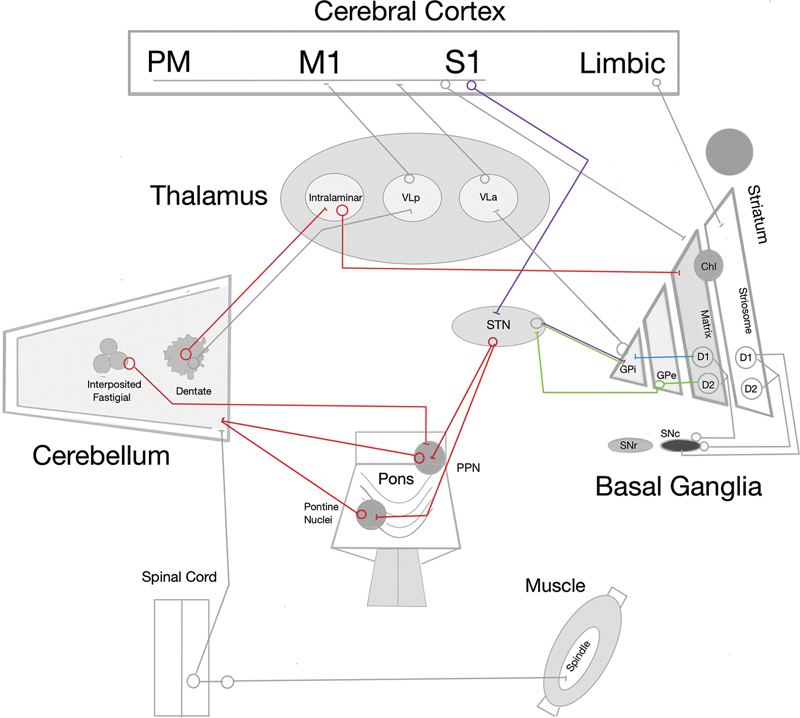
Peripheral lesions may contribute to the generation of dystonia through pathways involving the cerebellum.[16] [17] In 1924, Francis Martin Walshe (1885–1973) reported[18] that a diluted local anesthetic could improve akinesia in a patient with Parkinson's disease, reasoning that this effect was mediated by alterations in muscle afferent activity. Decades later, in 1995, Ryuji Kaji (1935–) injected diluted lidocaine into dystonic muscles and demonstrated that temporary blockade of muscle afferents could transiently suppress abnormal contractions.[17] When injected diffusely into a muscle, local anesthetics predominantly affect gamma efferent fibers that innervate muscle spindles. Because muscle afferents constitute the major input for kinesthetic sensation, and spindle afferent pathways project densely to the cerebellum via the spinocerebellar tracts, these findings underscore the importance of altered sensory feedback in motor control ([Figure 1]).[16] [19] [20] [21] Neuroimaging studies further support this concept, revealing structural and functional abnormalities in the cerebellum and its afferent pathways in patients with dystonia.[3] Taken together, these observations support the current hypothesis that dystonic movements and postures may be preprogrammed within subcortical circuits, even at rest, reflecting abnormal sensorimotor integration. Nowadays, although these pathways remain only partially understood, substantial anatomical evidence confirms the existence of direct disynaptic connections between the cerebellum and both the striatum and the subthalamic nucleus (STN, [Figure 1]).[16]
PATHOPHYSIOLOGY
Basal ganglia
Since the 1990s, the predominant clinical model of movement disorders involving the basal ganglia has proposed the existence of two primary opposing neural circuits, the direct and indirect pathways (commonly referred to as the Go and No-Go pathways).[22] According to this framework, balanced activity between these circuits is essential for normal motor control, whereas their imbalance results in either hypo- or hyperkinetic movement disorders. Nevertheless, controversies regarding the direct–indirect pathway model have steadily accumulated, and this framework has never adequately explained the pathophysiological origins of dystonia.[23]
According to the dual Go/No-Go model, D1 receptor-expressing striatal projection neurons (D1SPNs) that project to the globus pallidus internus (GPi) initiate the pallido-thalamocortical system (and the pallido-nigro-thalamocortical system), forming the direct pathway that facilitates movement. In contrast, those expressing D2 receptors (D2SPNs) to the globus pallidus externus (GPe), which projects to the STN, which, in turn, projects to the GPi, form the indirect pathway, which inhibits movement ([Figure 1]).[22] [23] Over the past 4 decades, however, this framework has undergone extensive revisions. Experimental studies[23] have shown that D1SPNs and D2SPNs can be simultaneously active and cooperate, suggesting roles in reinforcement-based learning rather than a strict Go/No-Go dichotomy.
In fully-learned motor tasks, the pathway affected in dystonia is classified as a reinforcement learning circuit within the basal ganglia mediated by the dopaminergic system. As motor skills and efficiency improve, there is a shift from supervised to reinforcement learning, with greater involvement of the basal ganglia.[19] [24] However, if excessive dopamine was the sole cause of dystonia, important clinical conditions would remain unexplained, such as tardive dystonia and dopa-responsive dystonia. Although atypical antipsychotic drugs are associated with a lower risk of tardive syndromes, a substantial number of psychiatric patients continue to develop dystonia while using dopaminergic antagonists. Conversely, patients with dopa-responsive dystonia show dramatic improvement with levodopa treatment.[19]
Therefore, reinforcement learning models have further reshaped the interpretation of basal ganglia function, demonstrating that stimulation of D1SPNs is reinforcing and stimulation of D2SPNs is aversive, with pathway roles shown to be conditional and experience-dependent. Anatomical discoveries have added additional complexity: the hyperdirect pathway provides a rapid bypass of striatal processing, and the GPe has emerged not as a passive relay but as a key regulatory hub that dynamically shapes basal ganglia output through its projections to the STN, GPi, and striatum ([Figure 1]). Cell-type–specific studies have shown that distinct GPe neuronal populations exert opposing influences on these downstream targets, enabling the GPe to fine-tune movement rather than merely transmit signals. This expanded view underscores the limitations of the classical model and reinforces the need for a more dynamic framework of basal ganglia circuitry [21].
A recent study[23] has revealed that the canonical direct–indirect model is echoed by a parallel organization arising from the striosome compartment of the striatum. Unlike the matrix-derived pathways, which project to motor-output nuclei, the striosomal direct (S-D1) and indirect (S-D2) pathways target dopamine-containing neurons of the substantia nigra pars compacta (SNc). The striosome compartment receives prominent inputs from limbic-related cortical regions, linking these circuits to motivation, mood, and decision-making ([Figure 1]). Remarkably, the striosomal pathways exert effects opposite to those of the canonical pathways: activation of S-D1 reduces movement and dopamine release, while activation of S-D2 increases movement. This parallel organization suggests that striosomes provide homeostatic modulation of basal ganglia output and may contribute to non-motor symptoms.[23] Remarkable histopathological loss of the striosome compartment in the striatum is found in the dystonic phase of X-linked dystonia-parkinsonism (XDP).[25] [26]
Thus, while the direct–indirect pathway model remains a valid starting point, its initial simplicity has proven insufficient to explain complex movement disorders such as dystonia. Contemporary understanding recognizes that basal ganglia circuits operate through dynamic cooperation between pathways, integration with reinforcement learning, and conditional and context-dependent functions, as well as critical contributions from structures such as the GPe and the hyperdirect pathway. These revisions underscore the strengths and limitations of the canonical model, highlighting the necessity of moving beyond the traditional Go/No-Go framework.[23]
Cerebellum and its pathways
Traditionally, the basal ganglia and the cerebellum were considered independent motor circuits that only communicated indirectly through cortical relays. However, anatomical and physiological evidence[16] have demonstrated the existence of direct disynaptic connections between these structures. Specifically, the dentate nucleus projects to the striatum via the intralaminar nuclei of the thalamus, while the STN provides disynaptic outputs to the cerebellar cortex through the pontine nuclei or pedunculopontine nucleus ([Figure 1]).[4] [19] [27] [28] [29] Electrophysiological studies[30] in awake mice have confirmed that these pathways enable the cerebellum to modulate striatal activity with short latency (∼ 10 ms).
In the intact system, cortical high-frequency stimulation typically induces long-term depression (LTD) at corticostriatal synapses. When cerebellar input is coactivated, however, synaptic plasticity shifts towards long-term potentiation (LTP).[30] This suggests that the cerebellum plays a crucial role in shaping corticostriatal plasticity, thereby influencing the integration of sensory, motor, and dopaminergic signals necessary for fine-tuned motor control.[19] Cholinergic interneurons (ChIs)—strategically positioned at the border between the striosome and matrix striatum compartments—appear to be key mediators of this regulation, as they modulate dopamine release through nicotinic and muscarinic receptors.[31] [32] In dystonia, this physiological balance is disrupted by aberrant cerebellar inputs.[19] Experimental models have shown[30] that abnormally high-frequency cerebellar signals bias corticostriatal synapses toward pathological LTP, impairing the ability to depotentiate. This maladaptive plasticity has been demonstrated in models of dopa-induced dyskinesia[33] and in patients with DYT1 (TOR1A) dystonia,[34] [35] suggesting a shared mechanism of abnormal synaptic reinforcement.
A recent study[36] using genetic mouse models and electrophysiological recordings has provided new clarity, particularly through the identification of disease-specific spike signatures in the interposed cerebellar nucleus. In carefully designed models, disruption of Purkinje cell Gamma-aminobutyric acid (GABA)-ergic transmission in Pcp2Cre and Slc32a1fl/fl mice reliably produced ataxia. By contrast, the selective elimination of glutamatergic climbing fiber input to Purkinje cells in Ptf1aCre and Slc17a6fl/fl mice resulted in twisting postures and limb hyperextension, consistent with dystonia.[36]
Recordings from the interposed nuclei have revealed that each disorder was associated with distinct neuronal firing patterns, or spike signatures.[36] Ataxia was associated with two distinct yet consistent signatures, whereas dystonia exhibited its own stereotyped patterns. Notably, the severity of dystonia correlated with the balance between normal and pathological activity: mice with mild, stress-triggered dystonia retained more neurons exhibiting control-like firing, whereas severe models, such as ouabain-infused or Ptf1aCre and Slc17a6fl/fl mice, showed a predominance of neurons with a dystonia-specific signature. These findings establish a direct link between the relative proportion of neurons expressing pathological activity in the interposed nucleus and the clinical severity of dystonia. Perhaps most compelling, optogenetic induction of these spike signatures in otherwise normal mice was sufficient to reproduce the characteristic motor behaviors of ataxia or dystonia. This demonstrates that the pattern of cerebellar output, rather than structural lesions, is the critical determinant of phenotype.[36]
While most attention has historically focused on cerebellar efferents from the dentate nucleus, these findings underscore the pivotal role of other cerebellar nuclei. Through its extensive connections with the pedunculopontine nuclei, reticular formation, and other brainstem centers, the interposed nucleus emerges as a central hub through which cerebellar dysfunction may diversify into distinct motor syndromes.
CLINICAL AND GENETIC INSIGHTS
Many genetic diseases display complex phenotypes resulting from neurodegeneration across multiple systems and structures, including the basal ganglia and the cerebellum. This dual involvement may help explain the frequent co-occurrence of dystonia in patients with ataxia and other neurological manifestations. Network-based approaches have also provided compelling evidence[3] [4] [37] [38] that the molecular pathways underlying ataxia and dystonia are closely interconnected.
Cerebellar ataxia and dystonia have distinct physiological correlates; yet, they share specific abnormalities in muscle activation patterns, including prolonged agonist activity and mistimed recruitment of antagonists. Cerebellar ataxia may arise from two different mechanisms: loss of cerebellar function or, conversely, an abnormal gain in function. In patients with ataxia attributed to loss of function, the pattern of agonist–antagonist activation differs from that observed in dystonia. However, in a subset of ataxic patients, the activation pattern overlaps with that of dystonia, suggesting that their ataxia may reflect an abnormal gain in cerebellar output. Such gain-in-function phenomena may also occur occasionally in the spinocerebellar ataxias, in which ataxia and dystonia coexist, and the most pronounced morphological changes are found in the cerebellum. Viewed in this way, progressively-increasing abnormal gain in cerebellar function may represent a continuum that includes dystonia, in which cerebellar output fails to adequately distinguish signals intended for agonist and antagonist muscles, thereby producing co-contraction and motor overflow.[39]
A critical role for the cerebellum in the pathophysiology of dystonia[4] [37] is also supported by multiple lines of clinical evidence, including lesion location in secondary dystonia,[40] [41] the syndrome of dystonia with cerebellar atrophy (DYTCA),[42] and postmortem pathological findings in cervical dystonia.[43] Moreover, dystonia may be a presenting or prominent feature in several autosomal dominant hereditary ataxias; however, it is essential to note that these are multisystemic diseases characterized by degeneration that is not confined to the cerebellum ([Table 1]).[4] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] [54] [55] [56] [57]
|
SCA |
Prevalence of dystonia |
Clinical features |
References |
|---|---|---|---|
|
SCA-ATXN1 SCA1 |
12–13% |
Associated with a greater severity of ataxia |
|
|
SCA-ATXN2 SCA2 |
14–18% |
Associated with a greater severity of ataxia |
|
|
SCA-ATXN3 SCA3-MJD |
∼ 24% |
Associated with a greater severity of ataxia |
|
|
SCA-CACNA1A SCA6 |
5–9% |
Slower progression; mainly cerebellar cortex involvement |
|
|
SCA-ATXN7 SCA7 |
Case reports |
Craniocervical dystonia; writer's cramp; cerebellar and brainstem atrophies |
|
|
SCA-ATXN8OS SCA8 |
Case report |
Oromandibular dystonia; cerebellar atrophy |
[48] |
|
SCA-PPP2R2B SCA12 |
Case report |
Cervical dystonia; dysphonia; tremor |
[49] |
|
SCA-PRKCG SCA14 |
∼ 32% |
Focal/task-specific dystonia; myoclonus is frequent |
[50] |
|
SCA-TBP SCA17 |
Frequent |
Chorea; multiple dystonias (blepharospasm, torticollis, writer's cramp, foot dystonia) |
[51] |
|
SCA-KCND3 SCA19/22 |
Rare cases |
Childhood onset with ataxia, dystonia, myoclonus, and cognitive impairment |
[52] |
|
SCA-TGM6 SCA35 |
∼ 12% |
Cranial dystonia; head tremor with DBS response |
|
|
SCA-NOP56 SCA36 |
Case reports |
Focal dystonia; dystonic tremor; late onset |
[55] |
|
SCA-STUB1 SCA48 |
Case reports |
Cognitive decline; focal dystonia; chorea; myoclonus |
[56] |
|
SCA-ATN1 DRPLA |
Case reports |
Focal/segmental dystonia; often with chorea and myoclonus |
[57] |
Abbreviations: CBS, deep brain stimulation; DRPLA, dentatorubral-pallidoluysian atrophy; MJD, Machado-Joseph disease; SCA, spinocerebellar ataxia.
Autosomal recessive cerebellar ataxias (ARCAs) also constitute a heterogeneous group in which dystonia may occur as part of the clinical phenotype. The most prevalent ARCAs associated with dystonia include Friedreich ataxia (FXN), ataxia-telangiectasia (ATM), ataxia with isolated vitamin E deficiency (TTPA), POLG-related disorders, ataxia with oculomotor apraxia types 1, 2, and 4 (APTX, SETX, and PNKP respectively), PRRT2-associated disorders, and autosomal recessive spinocerebellar ataxia type 5 or Galloway–Mowat syndrome (WDR73).[3]
When dystonia is the only presenting feature aside from a possible tremor, it is referred to as isolated dystonia. In contrast, dystonia associated with another movement disorder or as part of broader neurological or systemic syndromes is classified as combined dystonia. The first isolated dystonia gene, TOR1A, was identified almost 30 years ago. Since then, the number of known genetic forms has increased significantly, primarily due to the advancement in next-generation sequencing (NGS) technologies.[2] Importantly, some monogenic dystonias, both isolated and combined, may occasionally manifest with ataxia ([Table 2]).[58] [59] [60] [61] [62] [63]
|
Dystonia |
Phenotypic spectrum and clinical features |
References |
|---|---|---|
|
DYT-TUBB4A DYT4 |
TUBB4A encompasses a broad spectrum, ranging from DYT4 to hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) leukoencephalopathy, characterized by symptoms including dystonia, psychomotor delay, spasticity, ataxia, dysarthria, short stature, and microcephaly. DYT4 should not be categorized as an isolated dystonia. |
[58] |
|
DYT-THAP1 DYT6 |
Clinically heterogeneous. A reported case demonstrated dystonia followed by the subsequent development of ataxia; transcranial magnetic stimulation (TMS) revealed absent cerebellar inhibition (CBI), supporting cerebellar involvement in the phenotype. |
[59] |
|
MYC/DYT-SGCE DYT11 |
Classically, myoclonus-dystonia with childhood onset, but atypical presentations may include episodic ataxia and opsoclonus-myoclonus-like features. |
[60] |
|
DYT/PARK-ATP1A3 DYT12 |
ATP1A3 is associated with a broad neurological spectrum, including alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, cerebellar ataxia-areflexia-pes cavus-optic atrophy-sensorineural hearing loss (CAPOS) syndrome, relapsing encephalopathy with cerebellar ataxia (RECA), and even psychiatric syndromes. |
[61] |
|
DYT-ANO3 DYT24 |
Slowly-progressive dystonia with facial grimacing, eyelid spasms, unstable gait, and progressive dysarthria/ataxia. One reported patient had prior midbrain and cerebellar infarction, but dystonia/ataxia progressed independently. |
[62] |
|
DYT-KMT2B DYT28 |
Novel splice-site variant described in a patient with adult-onset ataxia, mild dystonia, neuropathy, seizures, and ophthalmological involvement. Highlights the potential role of KMT2B in hereditary ataxias beyond dystonia. |
[63] |
Neuroimaging studies[64] in DYT1 (TOR1A) support the view that dystonia is a network and neurodevelopmental disorder, highlighting the role of imaging in elucidating its pathophysiology. Carriers of the TOR1A guanine-adenine-guanine (GAG) deletion exhibit metabolic changes in specific brain regions, characterized by reduced GABA and D2 receptor availability in the striatum, as well as abnormalities in the cerebello-thalamocortical pathway. This circuit can be divided into proximal (cerebellothalamic) and distal (thalamocortical) components: abnormalities restricted to the proximal part have been demonstrated in manifesting carriers. They are linked to increased activation of the supplementary motor area (SMA). In contrast, combined proximal and distal abnormalities occur in non-manifesting carriers, thought to prevent the spread of abnormal impulses to the cortex and thereby protect against clinical dystonia. In addition, normal fractional anisotropy in the superior cerebellar peduncle has been observed[64] in a patient with the 216H polymorphism, a finding associated with preservation of normal cerebello-thalamocortical connectivity.
TREATMENT INSIGHTS
Pharmacological treatments
The traditional understanding has been that anticholinergics alleviate dystonia by correcting a striatal neurotransmitter imbalance, characterized by reduced dopaminergic and increased cholinergic activities.[65] However, studies[66] [67] have expanded this view, suggesting that their beneficial effects may also involve the modulation of cerebellar circuits.
Within the striatal matrix, giant aspiny interneurons—also known as ChIs—serve as an intrinsic source of acetylcholine (Ach), whereas the pedunculopontine nucleus provides extrinsic cholinergic input.[68] Cholinergic interneurons are strongly modulated by thalamostriatal innervation, particularly from the cerebellar dentate nucleus via the intralaminar thalamus, more so than by corticostriatal input. Repetitive thalamic stimulation increases ChI firing, linking cerebellar output to striatal cholinergic activity. Dysfunction of ChIs is thought to contribute to abnormal motor pattern selection, consistent with both striosomal and cerebellar hypotheses of dystonia pathogenesis. Significantly, ChI activation can trigger dopamine release by engaging presynaptic nicotinic receptors, while striosomal dysfunction facilitates additional dopamine release through striosome–SNc circuits ([Figure 1]). Together, these processes converge to produce dopamine excess and increased movement amplitude via facilitation of the direct pathway.[23] [29]
Cerebellar neurons dynamically adjust their firing frequency according to the activation of agonist or antagonist muscles during motor tasks, with distinct Purkinje cells and downstream nuclear neurons likely involved in the precise timing of this activity.[39] Acetylcholine also plays a critical role in cerebellar function, with dense cholinergic projections terminating in the granule cell layer, thereby exerting a significant influence on cerebellar processing and associated behaviors.[69] Fore et al.[69] demonstrated in vitro that ACh produces a prolonged inhibitory effect on Golgi cells via muscarinic receptor activation, thereby reducing the inhibitory drive onto granule cells. Simultaneously, muscarinic receptor activation on mossy fibers diminishes excitatory input to granule cells. This concurrent reduction in excitation and inhibition alters spike probability in a heterogeneous manner—enhancing excitability in some granule cells while suppressing it in others. Significantly, ACh preferentially increases the excitability of strongly-inhibited granule cells, supporting the concept that granule cell inhibition is stimulus-specific and essential for cerebellar learning. These findings suggest that cholinergic neuromodulation can selectively enhance learning for specific mossy fiber inputs depending on behavioral context or stimulus salience.[69] Thus, ACh may serve as a key regulator of cerebellar plasticity, modulating the gain in signal processing from granule cells through Purkinje cells to the deep cerebellar nuclei.[39] Nevertheless, the in-vivo mechanisms that govern cerebellar ACh release and its precise impact on synaptic and network dynamics remain incompletely understood.[69]
Neurosurgical treatment
Deep brain stimulation (DBS) has been applied to different targets in dystonia, including the GPi, the ventrointermediate nucleus (VIM), and the STN.[19] [70] The GPi remains the most established target, and it is generally considered the first choice.[19] [70] However, GPi-DBS and STN-DBS are safe and effective, producing substantial improvements in patients' quality of life.[70] However, the choice of the optimal target remains the subject of debate.[70]
The rationale for GPi-DBS is better established: by increasing GABAergic inhibition from the GPi to the ventrolateral thalamus, it reduces thalamocortical excitatory drive to the premotor cortex, which is characteristically hyperexcitable in dystonia ([Figure 1]). In contrast, the mechanisms underlying STN-DBS remain less clearly defined. Some authors[19] [39] have suggested that dystonia may involve an imbalance with relative predominance of the direct pathway, and that STN stimulation could restore basal ganglia output by engaging the indirect pathway. However, this interpretation is uncertain. Alternative hypotheses propose that STN-DBS may modulate sensorimotor integration through the activation of either orthodromic thalamocortical or antidromic hyperdirect pathways. A particularly compelling possibility is that the delayed clinical improvement often observed after STN-DBS reflects adaptive changes in disynaptic projections from the STN to the cerebellar cortex via the pontine nuclei or pedunculopontine nucleus, thereby influencing cerebello-thalamocortical circuits ([Figure 1]). This perspective emphasizes the broader network interactions underlying dystonia.[19] [39]
CONCLUSION
Although the pathophysiology of dystonia has not been fully elucidated yet, it appears to be complex and involves multiple brain structures and their interconnected pathways. After being dismissed for decades as purely psychogenic, dystonic movements were subsequently attributed to imbalances within basal ganglia circuits. However, with the accumulation of evidence from recent studies and advances in neuromodulation techniques, such as DBS, it has become evident that these mechanisms are far more intricate. Today, it is clear that the cerebellum, in conjunction with the basal ganglia, plays a crucial role in the development of dystonia. Future investigations will likely clarify these contributions in greater detail, as well as uncover the involvement of additional structures currently regarded as secondary players. A deeper understanding of these circuits and their neurotransmitters will be crucial for the development of novel therapeutic strategies.
Conflict of Interest
The authors have no conflict of interest to declare.
Authors' Contributions
Conceptualization: CHFC; Methodology: CHFC; Project administration: CHFC, HAGT; Supervision: CHFC, HAGT; Validation: CHFC, HAGT; Visualization: CHFC, HAGT; Writing – original draft: CHFC; Writing – review & editing: CHFC, HAGT.
Data Availability Statement
Data will be available upon request to the corresponding author.
Editor-in-Chief: Ayrton Roberto Massaro (ORCID: 0000-0003-2305-1073).
Associate Editor: Orlando Graziani Povoas Barsottini (ORCID: 0000-0002-0107-0831).
-
References
- 1 Camargo CH, Teive HA. Evolution of the concept of dystonia. Arq Neuropsiquiatr 2014; 72 (07) 559-561
- 2 Albanese A, Bhatia KP, Fung VSC. et al. Definition and Classification of Dystonia. Mov Disord 2025; 40 (07) 1248-1259
- 3 Jinnah HA, Hess EJ. A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum?. Neurology 2006; 67 (10) 1740-1741
- 4 Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 2008; 131 (Pt 9): 2499-2509
- 5 Oppenheim H. Über eine eigenartige Krampfkrankheit des kindlichen und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia musculorum deformans). Neurol Centrabl 1911; 30: 1090-1107
- 6 Hallett M. Functional Neurologic Disorder, La Lésion Dynamique: 2024 Wartenberg Lecture. Neurology 2024; 103 (11) e210051
- 7 Schwalbe MW. Eine eigntumliche tonische Krampfform mit hysterischen Symptomen. Berlin: Schade; 1908
- 8 Lanska DJ. Early Controversies over Athetosis: I. Clinical Features, Differentiation from other Movement Disorders, Associated Conditions, and Pathology. Tremor Other Hyperkinet Mov (N Y) 2013; ;3:tre-03-132-2918-1
- 9 Goetz CG, Chmura TA, Lanska DJ. History of dystonia: part 4 of the MDS-sponsored history of movement disorders exhibit, Barcelona, June, 2000. Mov Disord 2001; 16 (02) 339-345
- 10 Herz E. Dystonia: I. Historical review: analysis of dystonic symptoms and physiologic mechanisms involved. Arch Neurol Psychiatry 1944; 51 :(4) 305-318
- 11 Zeman W, Dyken P. Dystonia musculorum deformans. Clinical, genetic and pathoanatomical studies. Psychiatr Neurol Neurochir 1967; 70 (02) 77-121
- 12 Martinez AR, Faber I, Martins Jr CR. et al. Derek Denny-Brown: the man behind the ganglia. Arq Neuropsiquiatr 2017; 75 (02) 127-129
- 13 Marsden CD. Motor disorders in basal ganglia disease. Hum Neurobiol 1984; 2 (04) 245-250
- 14 Gowers WR. A manual of diseases of the nervous system. London: Churchill; 1888. .V. 2:659
- 15 Lenka A, Jankovic J. Peripherally-induced Movement Disorders: An Update. Tremor Other Hyperkinet Mov (N Y) 2023; 13: 8
- 16 Kaji R, Bhatia K, Graybiel AM. Pathogenesis of dystonia: is it of cerebellar or basal ganglia origin?. J Neurol Neurosurg Psychiatry 2018; 89 (05) 488-492
- 17 Kaji R, Rothwell JC, Katayama M. et al. Tonic vibration reflex and muscle afferent block in writer's cramp. Ann Neurol 1995; 38 (02) 155-162
- 18 Walshe FMR. Observations on the nature of the muscular rigidity of paralysis agitans, and on its relationship to tremor. Brain 1924; 47 (02) 159-177
- 19 Kaji R. Direct cerebello-striatal loop in dystonia as a possible new target for deep brain stimulation: A revised view of subcortical pathways involved. Front Neurol 2022; 13: 912818
- 20 Camargo CHF, Ferreira-Peruzzo SA, Ribas DIR, Franklin GL, Teive HAG. Imbalance and gait impairment in Parkinson's disease: discussing postural instability and ataxia. Neurol Sci 2024; 45 (04) 1377-1388
- 21 Wang AS, Alkhodair IM, Kilbane CW. The role of the cerebellum in dystonia. Dystonia 2025; 4: 14692
- 22 Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 1990; 13 (07) 266-271
- 23 Graybiel AM. Surprises From the Basal Ganglia: Stop and Go Have New Meaning. Mov Disord 2025; 40 (10) 2077-2082
- 24 Lehéricy S, Benali H, Van de Moortele PF. et al. Distinct basal ganglia territories are engaged in early and advanced motor sequence learning. Proc Natl Acad Sci U S A 2005; 102 (35) 12566-12571
- 25 Goto S, Lee LV, Munoz EL. et al. Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann Neurol 2005; 58 (01) 7-17
- 26 Hanssen H, Heldmann M, Prasuhn J. et al. Basal ganglia and cerebellar pathology in X-linked dystonia-parkinsonism. Brain 2018; 141 (10) 2995-3008
- 27 Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci U S A 2010; 107 (18) 8452-8456
- 28 Bostan AC, Strick PL. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci 2018; 19 (06) 338-350
- 29 Morigaki R, Miyamoto R, Matsuda T, Miyake K, Yamamoto N, Takagi Y. Dystonia and Cerebellum: From Bench to Bedside. Life (Basel) 2021; 11 (08) 776
- 30 Chen CH, Fremont R, Arteaga-Bracho EE, Khodakhah K. Short latency cerebellar modulation of the basal ganglia. Nat Neurosci 2014; 17 (12) 1767-1775
- 31 Cover KK, Gyawali U, Kerkhoff WG. et al. Activation of the rostral intralaminar thalamus drives reinforcement through striatal dopamine release. Cell Rep 2019; 26 (06) 1389-1398.e3
- 32 Abudukeyoumu N, Hernandez-Flores T, Garcia-Munoz M, Arbuthnott GW. Cholinergic modulation of striatal microcircuits. Eur J Neurosci 2019; 49 (05) 604-622
- 33 Picconi B, Centonze D, Håkansson K. et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci 2003; 6 (05) 501-506
- 34 Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci 2007; 30 (05) 211-219
- 35 Ruge D, Cif L, Limousin P. et al. Shaping reversibility? Long-term deep brain stimulation in dystonia: the relationship between effects on electrophysiology and clinical symptoms. Brain 2011; 134 (Pt 7): 2106-2115
- 36 Van der Heijden ME, Brown AM, Kizek DJ, Sillitoe RV. Cerebellar nuclei cells produce distinct pathogenic spike signatures in mouse models of ataxia, dystonia, and tremor. eLife 2024; 12: RP91483
- 37 Rossi M, Balint B, Millar Vernetti P, Bhatia KP, Merello M. Genetic Dystonia-ataxia Syndromes: Clinical Spectrum, Diagnostic Approach, and Treatment Options. Mov Disord Clin Pract 2018; 5 (04) 373-382
- 38 Shakkottai VG, Batla A, Bhatia K. et al. Current Opinions and Areas of Consensus on the Role of the Cerebellum in Dystonia. Cerebellum 2017; 16 (02) 577-594
- 39 Shakkottai VG. Physiologic changes associated with cerebellar dystonia. Cerebellum 2014; 13 (05) 637-644
- 40 LeDoux MS, Brady KA. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord 2003; 18 (01) 60-69
- 41 Waln O, LeDoux MS. Delayed-onset oromandibular dystonia after a cerebellar hemorrhagic stroke. Parkinsonism Relat Disord 2010; 16 (09) 623-625
- 42 Le Ber I, Clot F, Vercueil L. et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology 2006; 67 (10) 1769-1773
- 43 Prudente CN, Pardo CA, Xiao J. et al. Neuropathology of cervical dystonia. Exp Neurol 2013; 241: 95-104
- 44 Kuo PH, Gan SR, Wang J. et al. Dystonia and ataxia progression in spinocerebellar ataxias. Parkinsonism Relat Disord 2017; 45: 75-80
- 45 Schmitz-Hübsch T, Coudert M, Bauer P. et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology 2008; 71 (13) 982-989
- 46 Lin Y, Zheng JY, Jin YH, Xie YC, Jin ZB. Trinucleotide expansions in the SCA7 gene in a large family with spinocerebellar ataxia and craniocervical dystonia. Neurosci Lett 2008; 434 (02) 230-233
- 47 Gaillard N, Castelnovo G, Brice A, Labauge P. . [Writer's cramp secondary to spinocerebellar ataxia type 7.] Rev Neurol (Paris) 2007; 163 (05) 589-591
- 48 Ushe M, Perlmutter JS. Oromandibular and lingual dystonia associated with spinocerebellar ataxia type 8. Mov Disord 2012; 27 (14) 1741-1742
- 49 Neo S, Magrinelli F, Cordivari C, Bhatia KP. Tongue Protrusion and Feeding Dystonia Can Develop in PPP2R2B-Related Spinocerebellar Ataxia. Mov Disord Clin Pract 2024; 11 (05) 578-579
- 50 Schmitz-Hübsch T, Lux S, Bauer P. et al. Spinocerebellar ataxia type 14: refining clinicogenetic diagnosis in a rare adult-onset disorder. Ann Clin Transl Neurol 2021; 8 (04) 774-789
- 51 Rolfs A, Koeppen AH, Bauer I. et al. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol 2003; 54 (03) 367-375
- 52 Kurihara M, Ishiura H, Sasaki T. et al. Novel De Novo KCND3 Mutation in a Japanese Patient with Intellectual Disability, Cerebellar Ataxia, Myoclonus, and Dystonia. Cerebellum 2018; 17 (02) 237-242
- 53 Sharawat IK, Panda PK, Bhunia NS, Dawman L. Clinical Spectrum of TGM6-Related Movement Disorders: A New Report with a Pooled Analysis of 48 Patients. J Neurosci Rural Pract 2021; 12 (04) 656-665
- 54 Fasano A, Hodaie M, Munhoz RP, Rohani M. SCA 35 presenting as isolated treatment-resistant dystonic hand tremor. Parkinsonism Relat Disord 2017; 37: 118-119
- 55 Baviera-Muñoz R, Carretero-Vilarroig L, Muelas N. et al. Spinocerebellar Ataxia 36 is a Frequent Cause of Hereditary Ataxia in Eastern Spain. Mov Disord Clin Pract 2023; 10 (06) 992-997
- 56 Ravel JM, Benkirane M, Calmels N. et al. Expanding the clinical spectrum of STIP1 homology and U-box containing protein 1-associated ataxia. J Neurol 2021; 268 (05) 1927-1937
- 57 Hatano T, Okuma Y, Iijima M, Fujishima K, Goto K, Mizuno Y. Cervical dystonia in dentatorubral-pallidoluysian atrophy. Acta Neurol Scand 2003; 108 (04) 287-289
- 58 Vemula SR, Xiao J, Bastian RW, Momčilović D, Blitzer A, LeDoux MS. Pathogenic variants in TUBB4A are not found in primary dystonia. Neurology 2014; 82 (14) 1227-1230
- 59 Nikolov P, Hassan SS, Aytulun A. et al. Cerebellar Involvement in DYT-THAP1 Dystonia. Cerebellum 2019; 18 (05) 969-971
- 60 Drivenes B, Born AP, Ek J, Dunoe M, Uldall PV. A child with myoclonus-dystonia (DYT11) misdiagnosed as atypical opsoclonus myoclonus syndrome. Eur J Paediatr Neurol 2015; 19 (06) 719-721
- 61 Vezyroglou A, Akilapa R, Barwick K. et al. The Phenotypic Continuum of ATP1A3-Related Disorders. Neurology 2022; 99 (14) e1511-e1526
- 62 Fu F, Kang Y, Li J. et al. A Novel ANO3 Gene Mutation Associated with a Dystonia-Ataxia Syndrome. Mov Disord Clin Pract 2024; 11 (12) 1632-1634
- 63 Damásio J, Santos M, Samões R. et al. Novel KMT2B mutation causes cerebellar ataxia: Expanding the clinical phenotype. Clin Genet 2021; 100 (06) 743-747
- 64 Taiwo FT, Adebayo PB. Neuroimaging findings in DYT1 dystonia and the pathophysiological implication: A systematic review. Brain Behav 2023; 13 (06) e3023
- 65 Duvoisin RC. Cholinergic-anticholinergic antagonism in parkinsonism. Arch Neurol 1967; 17 (02) 124-136
- 66 Bohnen NI, Kanel P, Koeppe RA. et al. Regional cerebral cholinergic nerve terminal integrity and cardinal motor features in Parkinson's disease. Brain Commun 2021; 3 (02) fcab109
- 67 Jaarsma D, Ruigrok TJ, Caffé R. et al. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res 1997; 114: 67-96
- 68 Benarroch EE. Effects of acetylcholine in the striatum. Recent insights and therapeutic implications. Neurology 2012; 79 (03) 274-281
- 69 Fore TR, Taylor BN, Brunel N, Hull C. Acetylcholine Modulates Cerebellar Granule Cell Spiking by Regulating the Balance of Synaptic Excitation and Inhibition. J Neurosci 2020; 40 (14) 2882-2894
- 70 Fan H, Zheng Z, Yin Z, Zhang J, Lu G. Deep Brain Stimulation Treating Dystonia: A Systematic Review of Targets, Body Distributions and Etiology Classifications. Front Hum Neurosci 2021; 15: 757579
Address for correspondence
Publikationsverlauf
Eingereicht: 05. September 2025
Angenommen: 13. Oktober 2025
Artikel online veröffentlicht:
22. Dezember 2025
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution 4.0 International License, permitting copying and reproduction so long as the original work is given appropriate credit (https://creativecommons.org/licenses/by/4.0/)
Thieme Revinter Publicações Ltda.
Rua Rego Freitas, 175, loja 1, República, São Paulo, SP, CEP 01220-010, Brazil
Carlos Henrique Ferreira Camargo, Hélio Afonso Ghizoni Teive. The cerebellum in dystonia: key player or background support?. Arq Neuropsiquiatr 2025; 83: s00451814368.
DOI: 10.1055/s-0045-1814368
-
References
- 1 Camargo CH, Teive HA. Evolution of the concept of dystonia. Arq Neuropsiquiatr 2014; 72 (07) 559-561
- 2 Albanese A, Bhatia KP, Fung VSC. et al. Definition and Classification of Dystonia. Mov Disord 2025; 40 (07) 1248-1259
- 3 Jinnah HA, Hess EJ. A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum?. Neurology 2006; 67 (10) 1740-1741
- 4 Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 2008; 131 (Pt 9): 2499-2509
- 5 Oppenheim H. Über eine eigenartige Krampfkrankheit des kindlichen und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia musculorum deformans). Neurol Centrabl 1911; 30: 1090-1107
- 6 Hallett M. Functional Neurologic Disorder, La Lésion Dynamique: 2024 Wartenberg Lecture. Neurology 2024; 103 (11) e210051
- 7 Schwalbe MW. Eine eigntumliche tonische Krampfform mit hysterischen Symptomen. Berlin: Schade; 1908
- 8 Lanska DJ. Early Controversies over Athetosis: I. Clinical Features, Differentiation from other Movement Disorders, Associated Conditions, and Pathology. Tremor Other Hyperkinet Mov (N Y) 2013; ;3:tre-03-132-2918-1
- 9 Goetz CG, Chmura TA, Lanska DJ. History of dystonia: part 4 of the MDS-sponsored history of movement disorders exhibit, Barcelona, June, 2000. Mov Disord 2001; 16 (02) 339-345
- 10 Herz E. Dystonia: I. Historical review: analysis of dystonic symptoms and physiologic mechanisms involved. Arch Neurol Psychiatry 1944; 51 :(4) 305-318
- 11 Zeman W, Dyken P. Dystonia musculorum deformans. Clinical, genetic and pathoanatomical studies. Psychiatr Neurol Neurochir 1967; 70 (02) 77-121
- 12 Martinez AR, Faber I, Martins Jr CR. et al. Derek Denny-Brown: the man behind the ganglia. Arq Neuropsiquiatr 2017; 75 (02) 127-129
- 13 Marsden CD. Motor disorders in basal ganglia disease. Hum Neurobiol 1984; 2 (04) 245-250
- 14 Gowers WR. A manual of diseases of the nervous system. London: Churchill; 1888. .V. 2:659
- 15 Lenka A, Jankovic J. Peripherally-induced Movement Disorders: An Update. Tremor Other Hyperkinet Mov (N Y) 2023; 13: 8
- 16 Kaji R, Bhatia K, Graybiel AM. Pathogenesis of dystonia: is it of cerebellar or basal ganglia origin?. J Neurol Neurosurg Psychiatry 2018; 89 (05) 488-492
- 17 Kaji R, Rothwell JC, Katayama M. et al. Tonic vibration reflex and muscle afferent block in writer's cramp. Ann Neurol 1995; 38 (02) 155-162
- 18 Walshe FMR. Observations on the nature of the muscular rigidity of paralysis agitans, and on its relationship to tremor. Brain 1924; 47 (02) 159-177
- 19 Kaji R. Direct cerebello-striatal loop in dystonia as a possible new target for deep brain stimulation: A revised view of subcortical pathways involved. Front Neurol 2022; 13: 912818
- 20 Camargo CHF, Ferreira-Peruzzo SA, Ribas DIR, Franklin GL, Teive HAG. Imbalance and gait impairment in Parkinson's disease: discussing postural instability and ataxia. Neurol Sci 2024; 45 (04) 1377-1388
- 21 Wang AS, Alkhodair IM, Kilbane CW. The role of the cerebellum in dystonia. Dystonia 2025; 4: 14692
- 22 Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 1990; 13 (07) 266-271
- 23 Graybiel AM. Surprises From the Basal Ganglia: Stop and Go Have New Meaning. Mov Disord 2025; 40 (10) 2077-2082
- 24 Lehéricy S, Benali H, Van de Moortele PF. et al. Distinct basal ganglia territories are engaged in early and advanced motor sequence learning. Proc Natl Acad Sci U S A 2005; 102 (35) 12566-12571
- 25 Goto S, Lee LV, Munoz EL. et al. Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann Neurol 2005; 58 (01) 7-17
- 26 Hanssen H, Heldmann M, Prasuhn J. et al. Basal ganglia and cerebellar pathology in X-linked dystonia-parkinsonism. Brain 2018; 141 (10) 2995-3008
- 27 Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci U S A 2010; 107 (18) 8452-8456
- 28 Bostan AC, Strick PL. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci 2018; 19 (06) 338-350
- 29 Morigaki R, Miyamoto R, Matsuda T, Miyake K, Yamamoto N, Takagi Y. Dystonia and Cerebellum: From Bench to Bedside. Life (Basel) 2021; 11 (08) 776
- 30 Chen CH, Fremont R, Arteaga-Bracho EE, Khodakhah K. Short latency cerebellar modulation of the basal ganglia. Nat Neurosci 2014; 17 (12) 1767-1775
- 31 Cover KK, Gyawali U, Kerkhoff WG. et al. Activation of the rostral intralaminar thalamus drives reinforcement through striatal dopamine release. Cell Rep 2019; 26 (06) 1389-1398.e3
- 32 Abudukeyoumu N, Hernandez-Flores T, Garcia-Munoz M, Arbuthnott GW. Cholinergic modulation of striatal microcircuits. Eur J Neurosci 2019; 49 (05) 604-622
- 33 Picconi B, Centonze D, Håkansson K. et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci 2003; 6 (05) 501-506
- 34 Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci 2007; 30 (05) 211-219
- 35 Ruge D, Cif L, Limousin P. et al. Shaping reversibility? Long-term deep brain stimulation in dystonia: the relationship between effects on electrophysiology and clinical symptoms. Brain 2011; 134 (Pt 7): 2106-2115
- 36 Van der Heijden ME, Brown AM, Kizek DJ, Sillitoe RV. Cerebellar nuclei cells produce distinct pathogenic spike signatures in mouse models of ataxia, dystonia, and tremor. eLife 2024; 12: RP91483
- 37 Rossi M, Balint B, Millar Vernetti P, Bhatia KP, Merello M. Genetic Dystonia-ataxia Syndromes: Clinical Spectrum, Diagnostic Approach, and Treatment Options. Mov Disord Clin Pract 2018; 5 (04) 373-382
- 38 Shakkottai VG, Batla A, Bhatia K. et al. Current Opinions and Areas of Consensus on the Role of the Cerebellum in Dystonia. Cerebellum 2017; 16 (02) 577-594
- 39 Shakkottai VG. Physiologic changes associated with cerebellar dystonia. Cerebellum 2014; 13 (05) 637-644
- 40 LeDoux MS, Brady KA. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord 2003; 18 (01) 60-69
- 41 Waln O, LeDoux MS. Delayed-onset oromandibular dystonia after a cerebellar hemorrhagic stroke. Parkinsonism Relat Disord 2010; 16 (09) 623-625
- 42 Le Ber I, Clot F, Vercueil L. et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology 2006; 67 (10) 1769-1773
- 43 Prudente CN, Pardo CA, Xiao J. et al. Neuropathology of cervical dystonia. Exp Neurol 2013; 241: 95-104
- 44 Kuo PH, Gan SR, Wang J. et al. Dystonia and ataxia progression in spinocerebellar ataxias. Parkinsonism Relat Disord 2017; 45: 75-80
- 45 Schmitz-Hübsch T, Coudert M, Bauer P. et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology 2008; 71 (13) 982-989
- 46 Lin Y, Zheng JY, Jin YH, Xie YC, Jin ZB. Trinucleotide expansions in the SCA7 gene in a large family with spinocerebellar ataxia and craniocervical dystonia. Neurosci Lett 2008; 434 (02) 230-233
- 47 Gaillard N, Castelnovo G, Brice A, Labauge P. . [Writer's cramp secondary to spinocerebellar ataxia type 7.] Rev Neurol (Paris) 2007; 163 (05) 589-591
- 48 Ushe M, Perlmutter JS. Oromandibular and lingual dystonia associated with spinocerebellar ataxia type 8. Mov Disord 2012; 27 (14) 1741-1742
- 49 Neo S, Magrinelli F, Cordivari C, Bhatia KP. Tongue Protrusion and Feeding Dystonia Can Develop in PPP2R2B-Related Spinocerebellar Ataxia. Mov Disord Clin Pract 2024; 11 (05) 578-579
- 50 Schmitz-Hübsch T, Lux S, Bauer P. et al. Spinocerebellar ataxia type 14: refining clinicogenetic diagnosis in a rare adult-onset disorder. Ann Clin Transl Neurol 2021; 8 (04) 774-789
- 51 Rolfs A, Koeppen AH, Bauer I. et al. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol 2003; 54 (03) 367-375
- 52 Kurihara M, Ishiura H, Sasaki T. et al. Novel De Novo KCND3 Mutation in a Japanese Patient with Intellectual Disability, Cerebellar Ataxia, Myoclonus, and Dystonia. Cerebellum 2018; 17 (02) 237-242
- 53 Sharawat IK, Panda PK, Bhunia NS, Dawman L. Clinical Spectrum of TGM6-Related Movement Disorders: A New Report with a Pooled Analysis of 48 Patients. J Neurosci Rural Pract 2021; 12 (04) 656-665
- 54 Fasano A, Hodaie M, Munhoz RP, Rohani M. SCA 35 presenting as isolated treatment-resistant dystonic hand tremor. Parkinsonism Relat Disord 2017; 37: 118-119
- 55 Baviera-Muñoz R, Carretero-Vilarroig L, Muelas N. et al. Spinocerebellar Ataxia 36 is a Frequent Cause of Hereditary Ataxia in Eastern Spain. Mov Disord Clin Pract 2023; 10 (06) 992-997
- 56 Ravel JM, Benkirane M, Calmels N. et al. Expanding the clinical spectrum of STIP1 homology and U-box containing protein 1-associated ataxia. J Neurol 2021; 268 (05) 1927-1937
- 57 Hatano T, Okuma Y, Iijima M, Fujishima K, Goto K, Mizuno Y. Cervical dystonia in dentatorubral-pallidoluysian atrophy. Acta Neurol Scand 2003; 108 (04) 287-289
- 58 Vemula SR, Xiao J, Bastian RW, Momčilović D, Blitzer A, LeDoux MS. Pathogenic variants in TUBB4A are not found in primary dystonia. Neurology 2014; 82 (14) 1227-1230
- 59 Nikolov P, Hassan SS, Aytulun A. et al. Cerebellar Involvement in DYT-THAP1 Dystonia. Cerebellum 2019; 18 (05) 969-971
- 60 Drivenes B, Born AP, Ek J, Dunoe M, Uldall PV. A child with myoclonus-dystonia (DYT11) misdiagnosed as atypical opsoclonus myoclonus syndrome. Eur J Paediatr Neurol 2015; 19 (06) 719-721
- 61 Vezyroglou A, Akilapa R, Barwick K. et al. The Phenotypic Continuum of ATP1A3-Related Disorders. Neurology 2022; 99 (14) e1511-e1526
- 62 Fu F, Kang Y, Li J. et al. A Novel ANO3 Gene Mutation Associated with a Dystonia-Ataxia Syndrome. Mov Disord Clin Pract 2024; 11 (12) 1632-1634
- 63 Damásio J, Santos M, Samões R. et al. Novel KMT2B mutation causes cerebellar ataxia: Expanding the clinical phenotype. Clin Genet 2021; 100 (06) 743-747
- 64 Taiwo FT, Adebayo PB. Neuroimaging findings in DYT1 dystonia and the pathophysiological implication: A systematic review. Brain Behav 2023; 13 (06) e3023
- 65 Duvoisin RC. Cholinergic-anticholinergic antagonism in parkinsonism. Arch Neurol 1967; 17 (02) 124-136
- 66 Bohnen NI, Kanel P, Koeppe RA. et al. Regional cerebral cholinergic nerve terminal integrity and cardinal motor features in Parkinson's disease. Brain Commun 2021; 3 (02) fcab109
- 67 Jaarsma D, Ruigrok TJ, Caffé R. et al. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res 1997; 114: 67-96
- 68 Benarroch EE. Effects of acetylcholine in the striatum. Recent insights and therapeutic implications. Neurology 2012; 79 (03) 274-281
- 69 Fore TR, Taylor BN, Brunel N, Hull C. Acetylcholine Modulates Cerebellar Granule Cell Spiking by Regulating the Balance of Synaptic Excitation and Inhibition. J Neurosci 2020; 40 (14) 2882-2894
- 70 Fan H, Zheng Z, Yin Z, Zhang J, Lu G. Deep Brain Stimulation Treating Dystonia: A Systematic Review of Targets, Body Distributions and Etiology Classifications. Front Hum Neurosci 2021; 15: 757579