

Subscribe to RSS
DOI: 10.1055/s-0035-1561264
Organic Catalysts for Photocontrolled Polymerizations
Authors
Publication History
Received: 28 October 2015
Accepted: 24 November 2015
Publication Date:
21 January 2016 (online)

Abstract
Photocontrolled polymerization is an emerging field of study in which catalysis with organic-based dyes has enabled the practical and inexpensive synthesis of well-defined, spatially and temporally controlled polymeric structures. Herein, we review the current progress in the development of organic catalysts for photoregulated polymerizations. In particular, we highlight advances in metal-free variants of photomediated controlled radical polymerizations. In addition, we examine how the unique properties of these catalysts allow the development of two mechanistically distinct chain-growth polymerizations controlled by light.
1 Introduction
2 Photocontrolled Atom-Transfer Radical Polymerization (ATRP)
3 Photocontrolled Reversible Addition–Fragmentation Chain Transfer (RAFT)
4 Reversible Complexation-Mediated Polymerization (RCMP)
5 Photocontrolled Ring-Opening Metathesis Polymerization (ROMP)
6 Conclusion and Future Outlook
Biographical Sketches

Jacob T. Trotta was born in Peabody, Massachusetts. He studied chemistry at Villanova University and received his B. S. in 2014. Since then, he has been a Ph.D. student in the Fors group where he is working on the development of photocontrolled polymerization reactions.

Brett P. Fors was born in Polson, Montana and carried out his undergraduate studies in chemistry at Montana State University (B.S., 2006). He went on to do his Ph.D. (2011) at the Massachusetts Institute of Technology with Professor Stephen L. Buchwald. After his doctoral studies, he became an Elings Fellow at the University of California, Santa Barbara working with Professor Craig J. Hawker. In 2014, he joined the faculty at Cornell University and is currently an assistant professor in the Department of Chemistry and Chemical Biology.
Introduction
Polymers are essential and ubiquitous materials of everyday life, and the evolution of new chemical techniques to control polymer architecture leads to novel functional materials and new technologies. Photocontrolled polymerization is a burgeoning research area that offers great promise for delivering the capacity to tailor material properties through precise spatiotemporal regulation of polymer chain growth.[1] [2] The development of photoactive metal-based catalysts (e.g., Ir,[3] Cu,[4,5] Ru,[6] Fe,[7]) has dominated this field and has led to several radical polymerization methods that give exceptional control over polymerization kinetics and enables the formation of previously inaccessible structures.[7] [8]
Although these methods have revolutionized photopolymerizations, the use of metal-based catalysts can be prohibitively expensive, which restricts their practicality. In addition, the contamination of final materials with even trace quantities of metal can limit the use of these methods, especially in electronic or biomaterials applications.[9] [10] Although strategies have been developed to remove impurities from polymers,[11] they can be time-consuming and costly. These drawbacks have spawned investigations into the development of organic-based catalysts for photocontrolled, metal-free polymerizations.
Organic photoredox catalysts have seen widespread adoption in organic transformations.[12] [13] They have been used as drop-in surrogates for metal-based catalysts,[14,15] and in certain cases have provided unique reactivity and led to the development of new synthetic methods. This review focuses on recently developed organic catalysts for controlled polymerization reactions that allow photoregulation of polymer chain growth.[16] Specifically, we examine the development of metal-free variants of photocontrolled atom-transfer radical polymerization (ATRP) and photoinduced electron transfer-reversible addition–fragmentation chain transfer polymerization (PET-RAFT). In addition, we highlight how the unique reactivity of these organic catalysts has enabled the emergence of photocontrolled reversible complexation-mediated polymerization (RCMP) and photocontrolled ring-opening metathesis polymerization (ROMP) (Scheme [1]).

2
Photocontrolled Atom-Transfer Radical Polymerization (ATRP)
ATRP is one of the most widely used controlled radical polymerization (CRP) techniques. It operates through a redox equilibrium of ligated Cu, Ru, or Fe complexes with halide-capped polymer chains (Scheme [2]).[17] Advances in this technique have led to highly efficient protocols that give well-controlled polymerizations with living characteristics. Importantly, the ease of use and excellent monomer scope of these reactions enables the synthesis of a wide array of functional materials for a number of applications. Recently, there has been a surge in the development of photocontrolled ATRP-like processes that allow excellent spatial and temporal regulation of polymer chain growth.[2] However, metal contamination in the final materials and the requirement of expensive catalysts (e.g., Ir) remain limiting factors for both traditional and photocontrolled ATRP processes.

Hawker and co-workers[9] addressed these challenges by developing a photocontrolled ATRP process mediated by light and catalyzed by an organic catalyst. They had previously described a photoregulated ATRP process that used the photoredox catalyst fac-Ir(ppy)3 and proceeded through the proposed mechanism shown in Scheme [3].[3] [18] Building on this work, they sought a metal-free variant of Ir(ppy)3 that would operate through a similar mechanism and, ideally, offer the same control over the polymerization. This organic catalyst also required a highly reducing excited state similar to that of Ir(ppy)3 [–1.7 eV, Ir(IV)/Ir(III)*; see Scheme [3]]. With an excited state redox potential of –2.1 eV, 10-phenylphenothiazine (PTH) was found to be a suitable candidate for these polymerizations.

Using 0.1 mol% of PTH, the authors efficiently polymerized methyl methacrylate (MMA) via irradiation with 380 nm light (Scheme [4]). Significantly, this example was the first of a metal-free ATRP process. Other organic photoredox catalysts such as eosin Y and methylene blue have shown no polymerization under similar conditions, an outcome attributed to their oxidizing excited states and thus inability to activate the alkyl bromide chain end.[19] This observation demonstrated that a highly reducing organocatalyst such as PTH is needed for photocontrolled ATRP reactions.

This new PTH-based system efficiently produced poly(methyl methacrylate) (PMMA) samples with narrow dispersity (Đ) values. Đ is the ratio of weight-average (M w) to number-average (M n) molecular weights. Additionally, good agreements between experimental and theoretical M n values were observed. Similar to Ir-catalyzed photocontrolled ATRP, the PTH polymerization could effectively be turned on and off by cycling the exposure of the reaction to light; no background reaction was observed in the dark, and polymerization recommenced immediately after re-exposure to light (Figure [1a]). Importantly for these cycling studies, a linear relationship between M n and conversion was observed (Figure [1b]), as were first-order kinetics (Figure [1c]). These results demonstrated that the polymer chain ends were not irreversibly terminating in the absence of light and showed that similar to traditional ATRP, this polymerization protocol had living characteristics.
For a further demonstration of the living characteristics of this PTH system, chain-end fidelity was evaluated. A low-molecular-weight PMMA polymer was synthesized under PTH conditions and analyzed with electrospray ionization mass spectrometry (ESIMS) and 1H NMR spectroscopy. The results of ESIMS analysis showed a correlation between observed molecular weight and the expected values for the individual PMMA oligomers, which were capped with the desired alkyl bromide.
Although ESIMS data gave evidence for chain-end fidelity, chain extension studies gave definitive proof that a CRP system had been developed. With PMMA as a macroinitiator, a benzyl methacrylate (BnMA) block was grown to yield PMMA-b-PBnMA, in which the results of size exclusion chromatography (SEC) showed a clear shift to higher molecular weight without any observed tailing. This excellent retention of the alkyl bromide chain end in the PTH system further allowed chain extension with both Ir-catalyzed and traditional CuBr-based ATRP polymerizations that used methyl acrylate and styrene, respectively. This result shows that the PTH system is complementary to other metal-based ATRP methods.

Although the use of PTH as an organocatalyst has been critical in eliminating metal contamination in polymer products, Hawker[9] further showed that the utility of this catalyst goes beyond its metal surrogacy by demonstrating that its unique reactivity enables new chemistry. Specifically, for the polymerization of 2-(dimethylamino)ethyl methacrylate (DMAEMA), the use of Ir(ppy)3 resulted in polymers with broad Đ values and poor control over M n, a likely result of amine oxidation by the excited catalyst.[20] By contrast, PTH, which is more reducing (or less oxidizing) in the excited state than Ir(ppy)3, yielded poly[2-(dimethylamino)ethyl methacrylate] with good control and narrow Đ values (Scheme [4]). This outcome demonstrated an improvement in monomer scope for these photocontrolled ATRP processes and laid the groundwork for what could be future transformations that take advantage of the high reduction potential of this catalyst.
After this work by Hawker, Matyjaszewski and co-workers[10] used the PTH-catalyzed photocontrolled ATRP system for the polymerization of acrylonitriles. Under the optimized conditions, polyacrylonitrile was efficiently synthesized with good control over M n and Đ values of ~1.5. Matyjaszewski hypothesized that the broad Đ values could indicate slow deactivation due to low deactivator concentration or rate constants. Despite these broader distributions, good chain-end fidelity of polyacrylonitrile was demonstrated through chain extensions with an MMA macroinitiator for which SEC analysis showed a clear shift to higher molecular weight without any apparent tailing.
Matyjaszewski also explored the variation of N-aryl derivatives of PTH for these polymerizations (Scheme [5]), which showed activities comparable to those of the parent catalyst. Although PTH gave the best results, this key exploration of structure set the stage for future generations of efficient phenothiazine-derived catalysts.

Concurrently with Hawker’s original report, Miyake and Theriot[21] disclosed an ATRP protocol that used perylene as an organic photoredox catalyst (Scheme [6]). Using 0.1 mol% of perylene under white light irradiation, the authors synthesized PMMA and poly(n-butyl acrylate) with varying Đ values (1.29 to 1.85) and moderate control over M n. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry analysis demonstrated that a large number of the resulting polymers had alkyl bromide chain ends, however, it also showed that termination through radical–radical coupling and hydrogen abstraction were competitive processes. Alkyl bromide fidelity was further explored through the formation of block copolymers with a PMMA macroinitiator. Chain extensions with n-butyl methacrylate, butyl acrylate, styrene, and MMA were achieved, with some tailing observed in all cases.

These polymerizations with perylene were photoregulated, with little to no polymerization observed in the dark and first-order kinetics under light exposure (Figure [2]). M w, however, did not grow linearly with conversion, and Đ increased as the reaction proceeded. Miyake[21] hypothesized that these outcomes were a consequence of the low initiator efficiencies in these reactions. Although full control over polymerization was not observed for the perylene-based system, these reactions laid the groundwork for further discoveries with perylene-like catalysts.

Three systems have been developed in the pursuit of photocontrolled ATRP processes using organic photoredox catalysts. Importantly, these reports were the first of metal-free ATRP, which overcomes the long-standing issue of metal contamination in the final polymers and should enable new applications of these ATRP methods. Moreover, we anticipate that further exploration of these systems will provide access to a wider monomer scope and open the door for the synthesis of architecturally distinct polymers with precisely controlled properties.
3
Photocontrolled Reversible Addition–Fragmentation Chain Transfer (RAFT)
Similar to ATRP, RAFT is a widely used CRP method with living characteristics that enables the polymerization of a range of monomers.[22] RAFT protocols use chain transfer agents (CTAs) such as dithioesters, thiocarbamates, xanthates, and trithiocarbonates that enable degenerative chain transfer after radical initiation to give well-controlled polymerization processes (Scheme [7a]). In contrast to ATRP, RAFT does not require a catalyst; instead, a thermal initiator or photoinitiator such as 2,2′-azobis(isobutyronitrile) (AIBN) is used to establish degenerative chain transfer. To achieve spatiotemporal control over chain growth, researchers have recently focused on the use of photoredox catalysts to initiate polymerization reversibly (Scheme [7b]).


Efforts to control RAFT through photoredox chemistry (PET-RAFT) have been pioneered by Boyer and co-workers. Their original systems used Ir and Ru photoredox catalysts for reversible initiation of the RAFT process (Scheme [7b]).[23] [24] The proposed PET-RAFT mechanism was similar to that of photocontrolled ATRP, in which an excited photoredox catalyst reduces the CTA chain end to give the propagating radical, which can then undergo the degenerative chain transfer process (Scheme [8]). These systems allowed excellent photoregulation of polymerization, however, the use of a non-metal catalyst was desirable for reasons similar to those discussed for photo-ATRP (vide supra).

Boyer and co-workers[25] reported the use of the organic dye, eosin Y, as an efficient catalyst for these PET-RAFT processes (Scheme [8]). Eosin Y was chosen for its excited state redox potential that efficiently reduces the CTA to give the desired alkyl radical.[19] Moreover, because CTAs can themselves degrade under UV light, the use of an organocatalyst that absorbs light in the visible region was advantageous.[26] [27] Using 0.01 mol% of eosin Y (Scheme [8]) irradiated under 461 nm light, Boyer and colleagues polymerized MMA and multiple methacrylate derivatives (Figure [3]) to obtain polymers with narrow Đ values and good agreement between observed and theoretical M n’s. Importantly, the eosin Y system showed improved monomer scope compared with that of the Ir-based catalysts (specifically, DMAEMA and glycidyl methacrylate) previously used for these PET-RAFT polymerizations. Linear growth between M n and conversion as well as first-order kinetics and photocontrol were demonstrated for this method. Furthermore, chain-end fidelity was illustrated through efficient chain extensions of both MMA and oligoethylene glycol methyl ether methacrylate with a PMMA macroinitiator.
In addition to having a broad monomer scope and living characteristics, this catalytic system worked in the presence of oxygen. Importantly, good control was maintained when the reaction was exposed to oxygen, however, the reaction rates were slower than those of the anaerobic system. Boyer proposed that these reduced rates were due to oxygen quenching of the catalyst excited state, a well-known pathway for these photocatalysts.[28]
Boyer and co-workers performed the first PET-RAFT polymerizations under metal-free conditions with eosin Y as an organic catalyst. This approach enabled the visible-light-controlled synthesis of well-defined polymers from a variety of functionally diverse monomers, even in the presence of oxygen.
To increase the efficiency of the electron transfer process in these photocontrolled RAFT polymerizations, Boyer and colleagues[29] developed CTA initiators that were covalently bound to the organic dye, tetraphenylporphyrin (TPP). They reasoned that this strategy would increase the effective concentration of the catalyst relative to that of the CTA and improve the efficiency of the photoactivation, thereby leading to the traditional RAFT process (Scheme [9]). The use of 0.01 mol% of the tethered photocatalyst, TPP–3-benzylsulfanylthiocarbonylthiosulfanyl propionic acid (TPP–BSTP) with additional external CTA and 2-(n-butyltrithiocarbonate)propionic acid (BTPA) resulted in 56% conversion of MMA into a well-defined polymer (Figure [4]). Interestingly, in the control reaction in which TPP was not covalently bound to the CTA, only 3% conversion of MMA was detected after the same reaction time. These results corroborated Boyer’s hypothesis and illustrated that the electron transfer process was more efficient with the covalently bound catalyst/trithiocarbonate (TTC) system.
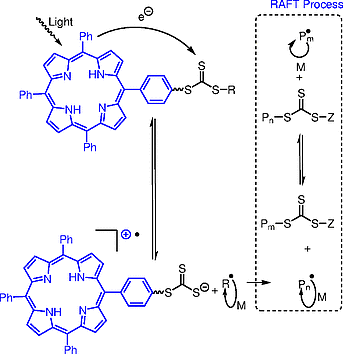
In further support of their postulate, Boyer and colleagues showed that the addition of linkers between the porphyrin and the CTA (TPP–C2–BSTP) (Figure [4]) resulted in reaction rates slower than those with TPP–BSTP. This result was attributed to the limitation of electron transfer by the increased distance between the TPP and CTA species. Furthermore, the synthesis of TPP–C2–2-(n-butyltrithiocarbonate)propionic acid, in which fragmentation and polymerization occurred between the catalyst and the CTA, resulted in even slower reaction rates. In this example, the distance between the CTA and TPP grew as polymerization proceeded, which decreased the electron transfer efficiency. Together the results of these control experiments elegantly show that the high activity of TPP–BSTP is due to an efficient intramolecular electron transfer process.
The end-group fidelity of these polymerizations was evaluated with chain extension experiments. Conveniently, no additional catalyst was required for these reactions because the photocatalyst was present at the chain end. Poly(methyl acrylate)-b-poly(N,N-dimethylamide) diblock copolymers were efficiently formed with this method.
This study by Boyer[29] shows that interaction between the CTA and the photoredox catalyst is necessary for efficient photocontrolled RAFT polymerization. This knowledge could enable the design of more effective systems that allow better control of the synthesis of varying polymer architectures.

Complementary to Boyer’s work,[23] [25] [30] and as an extension from their original work[31] with UV-activated TTC initiators, Johnson and co-workers[32] developed a visible-light-controlled RAFT process based on bifunctional TTC iniferters and catalyzed by PTH (Scheme [10]). With 0.4 mol% of TTC and 0.02 mol% of PTH, N-isopropyl acrylamide was polymerized to give polymers with narrow Đ values and observed M n’s matching theoretical data. M n also increased linearly with conversion, following first-order kinetics when illuminated with light. These conditions also produced high-molecular-weight polymers up to 159 kg/mol that were not achievable with a previous method Johnson had developed using UV light. These results illustrate that PTH is an effective catalyst for multiple photocontrolled CRPs.

Johnson further demonstrated chain-end fidelity in this system through extensions with poly(N,N-dimethylamide) macroinitiators that yielded triblock copolymers (Scheme [10]). Extensions were performed via three methods: (1) the organocatalyzed process discussed herein with N-isopropyl acrylamide, (2) UV irradiation to activate the macro-TTC initiator with ethoxy methyl ether acrylate as the monomer, and (3) traditional RAFT with AIBN as an initiator with t-butyl acrylate. This approach demonstrated that this method[32] was complementary to other RAFT processes and showed that it could be used to make well-defined functional materials.
Multiple organocatalyzed RAFT processes that are truly photoregulated have been developed and give excellent control over final polymer structures. Similar to organocatalyzed photocontrolled ATRP, these processes are likely to see further development and widespread use.
4
Reversible Complexation-Mediated Polymerization (RCMP)
Whereas ATRP and RAFT are well-established methods, RCMP, recently developed by Kaji and co-workers,[28] [33] [34] is a new CRP method that uses photoactive organic catalysts. RCMP is similar in mechanism to iodine-transfer polymerization, although it gives significantly improved control through its distinct activation pathway and non-reliance on degenerative chain transfer.[2,35] Kaji and colleagues originally published a method that used tributylamine as the catalyst and an alkyl iodide as the initiator for the polymerization of MMA (Scheme [11]).[36] They proposed that the amine and alkyl iodide formed a complex through halogen bonding that in turn caused homolytic C–I bond cleavage upon UV irradiation to give the propagating radical. Compared with traditional iodine transfer polymerization, this reversible activation resulted in PMMA with significantly narrower Đ values (~1.2) and better control over M n. Furthermore, these processes were efficiently regulated with light and did not require the use of metal-based catalysts.
More recently, Kaji and co-workers[37] reported a similar method with various catalysts with absorption maxima within the red to near-UV region of the electromagnetic spectrum (Scheme [11]). This method enabled efficient photocontrolled polymerizations in which the wavelength of light used in the reaction could be selected by choosing a specific catalyst. In contrast to photo-ATRP and PET-RAFT, which proceed through electron transfer mechanisms, these reactions reportedly used energy transfer processes exclusively to allow for controlled polymerization.
In all cases, the authors disclosed well-behaved reactions yielding polymers with narrow Đ values and controlled molecular weights. Moreover, the polymerization scope for this method was quite broad, with suitable monomers found in a wide array of functional methacrylate derivatives (Figure [5]). Furthermore, the chain-end fidelity of the system was evaluated through elemental analysis and suggested that 99% and 91% of the polymers were capped with iodine at 30% and 81% conversion, respectively. Additionally, these data were corroborated through 1H NMR analysis with an 800 MHz spectrometer, which showed ~99% chain-end fidelity at 30% conversion. Block copolymer synthesis with PMMA macroinitiators was also performed, and SEC analysis showed clear shifts to higher M n values. The controlled kinetics and good chain-end fidelity of this RCMP process broadens the scope of organocatalyzed photoredox polymerizations in general.


The ability to select a specific wavelength of light for these polymerizations will likely be highly advantageous. For example, it will enable the polymerization of monomers sensitive to photodegradation. Moreover, this capability has the potential to permit the temporal control of two different photomediated processes in a single reaction vessel through the appropriate selection of wavelength. Kaji and co-workers illustrated this concept with a one-pot block copolymer formation that relied on two different light-promoted reactions. With an alkyl iodide initiator functionalized with a pendant alcohol, they used 602 nm light to synthesize a PMMA block with RCMP. The irradiation wavelength was then adjusted to 350 nm and a poly(valerolactone) block was grown from the free alcohol with a photoacid generator as the catalyst (Scheme [12]).[37] [38] This example shows that appropriate wavelength selection can give temporal control over separate reactions in the same vessel.

In a manner similar to that of RCMP with alkyl iodides, Qiao reported[39] a process using tertiary amines to photocatalytically activate CTAs for RAFT polymerizations (Scheme [13]). The polymers obtained with this method exhibited narrow Đ values (1.26 to 1.31), and the approach offered good control over molecular weight. Polymerization did not occur without irradiation, and uncontrolled polymerization was observed when the reaction was carried out in the absence of the tertiary amine catalyst. The authors proposed that this outcome occurred owing to the degradation of the CTA upon UV irradiation. This report combined RCMP and PET-RAFT in a photocontrolled polymerization process.


5
Photocontrolled Ring-Opening Metathesis Polymerization (ROMP)
ROMP is a living chain-growth process that typically uses Ru- or Mo-based catalysts for efficient polymerization of cyclic olefins (Scheme [14]).[40] [41] The excellent control and broad functional group tolerance of this method has facilitated its use in a number of applications. Recently, Boydston and co-workers[42] developed a method for photocontrolling these ROMP processes through the use of an organic-based photoredox catalyst.
Unlike the other methods that we have discussed thus far, traditional ROMP does not proceed through a radical mechanism, which makes it difficult to envisage the use of a photoredox catalyst to promote these reactions. The approach to metal-free photocontrolled ROMP by Boydston and colleagues resulted in a mechanism switch to initiate a radical process. With a pyrylium salt as the photocatalyst and a vinyl ether as the initiator, norbornene was efficiently polymerized under irradiation with blue light. They hypothesized that the excited photocatalyst oxidized the vinyl ether to give a radical cation that underwent a [2+2] cycloaddition with the norbornene monomer (Scheme [15]). Fragmentation of this intermediate to relieve ring strain yielded a cyclopentane appended with an oxidized vinyl ether. This outcome enables further [2+2]/fragmentation cycles to give a chain growth polymerization. Importantly, the photocatalyst reversibly activated the vinyl ether chain end to give rise to a controlled photoregulated process.

The optimized conditions of the Boydston and co-workers approach yielded polymers with Đ values ranging from 1.3 to 1.7, and the final molecular weight could be relatively well-controlled by varying the stoichiometry of vinyl ether to norbornene. Over the course of polymerization, an increase in M n with conversion was observed, verifying the chain growth nature of this photoregulated ROMP. Although the collected data showed that the photoregulated ROMP was less controlled than traditional metal-catalyzed processes, the authors indicated that the poly(norbornene) polymers prepared with both methods had similar glass-transition temperatures, which indicated that the physical properties of the polymers were not significantly different. Additionally, temporal control of polymer chain growth was demonstrated by cycling the exposure of the reactions to visible light; no polymerization was observed in the dark.
This work by Boydston and colleagues is the first example of metal-free ROMP and achieves a radical process catalyzed by a photoactive catalyst. Notably, a recent study reported that this reaction can be used to synthesize metal-free cross-linked networks that are difficult to access through traditional means.[43] The photocontrol of this method also allows the spatiotemporal control of cyclic olefin polymerization, and we anticipate that this process will enable future applications of ROMP.
6
Conclusion and Future Outlook
Organic-based photoredox catalysts offer practical and cost-effective alternatives to their metal-based counterparts, and they display distinctive and interesting reactivities that can lead to new transformations. This review discusses metal-free catalysts that have been successfully developed for photocontrolled ATRP and PET-RAFT. In addition to being highly efficient for these reactions, these catalysts provide alternative reactivity that has broadened the substrate scope for both types of polymerization. Furthermore, organic catalysts have led the way to the development of photocontrolled RCMP and ROMP, both of which were previously inaccessible. We anticipate that further advances in this field will increase the practicality of these reactions and lead to new types of polymerization. Altogether, research in this area will further our ability to synthesize functional materials and lead to new discoveries.
Acknowledgment
The authors thank Cornell University for support.
-
References and Notes
- 1 Leibfarth FA, Mattson KM, Fors BP, Collins HA, Hawker CJ. Angew. Chem. Int. Ed. 2013; 52: 199
- 2 Yamago S, Nakamura Y. Polymer 2013; 54: 981
- 3 Fors BP, Hawker CJ. Angew. Chem. Int. Ed. 2012; 51: 8850
- 4 Mosnáček J, Ilčíková M. Macromolecules 2012; 45: 5859
- 5 Tasdelen MA, Uygun M, Yagci Y. Macromol. Chem. Phys. 2010; 211: 2271
- 6 Alfred NV, Jalapa NE, Morales SL, Ryabov AD, Le Lagadec R, Alexandrova L. Macromolecules 2012; 45: 8135
- 7 Telitel S, Dumur F, Campolo D, Poly J, Gigmes D, Fouassier JP, Lalevée J. J. Polym. Sci., Part A: Polym. Chem. 2015; DOI: 10.1002/pola.27896
- 8 Zhang T, Chen T, Amin I, Jordan R. Polym. Chem. 2014; 5: 4790
- 9 Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, de Alaniz JR, Fors BP, Hawker CJ. J. Am. Chem. Soc. 2014; 136: 16096
- 10 Pan X, Lamson M, Yan J, Matyjaszewski K. ACS Macro Lett. 2015; 4: 192
- 11 Matyjaszewski K, Pintauer T, Gaynor S. Macromolecules 2000; 33: 1476
- 12 Nicewicz DA, Nguyen TM. ACS Catal. 2014; 4: 355
- 13 Neumann M, Füldner S, König B, Zeitler K. Angew. Chem. Int. Ed. 2011; 50: 951
- 14 Hamilton DS, Nicewicz DA. J. Am. Chem. Soc. 2012; 134: 18577
- 15 Nguyen T, Nicewicz DA. J. Am. Chem. Soc. 2013; 135: 9588
- 16a Perkowski AJ, You W, Nicewicz DA. J. Am. Chem. Soc. 2015; 137: 7580
- 16b Xiao P, Zhang J, Dumur F, Tehfe MA, Morlet-Savary F, Graff B, Gigmes D, Fouassier JP, Lalevée J. Prog. Polym. Sci. 2015; 41: 32
- 17 Pintauer T, Matyjaszewski K. Chem. Soc. Rev. 2008; 37: 1087
- 18 Treat NJ, Fors BP, Kramer JW, Christianson M, Chiu CY, de Alaniz JR, Hawker CJ. ACS Macro Lett. 2014; 3: 580
- 19 Majek M, Filace F, von Wangelin AJ. Beilstein J. Org. Chem. 2014; 10: 981
- 20 Nguyen JD, D’Amato EM, Narayanam JM. R, Stephenson CR. J. Nat. Chem. 2012; 4: 854
- 21 Miyake GM, Theriot JC. Macromolecules 2014; 47: 8255
- 22 Moad G, Rizzardo E, Thang SH. Polymer 2008; 49: 1079
- 23 Xu J, Jung K, Atme A, Shanmugam S, Boyer C. J. Am. Chem. Soc. 2014; 136: 5508
- 24 Fu C, Xu J, Tao L, Boyer C. ACS Macro Lett. 2014; 3: 633
- 25 Xu J, Shanmugam S, Duong HT, Boyer C. Polym. Chem. 2015; 6: 5615
- 26 Quinn J, Barner L, Barner-Kowollik C, Rizzardo E, Davis TP. Macromolecules 2002; 35: 7620
- 27 Wang H, Li Q, Dai J, Du F, Zheng H, Bai R. Macromolecules 2013; 46: 2576
- 28 Goto A, Ohtsuki A, Ohfuji H, Tanishima M, Kaji H. J. Am. Chem. Soc. 2013; 135: 13286
- 29 Xu J, Shanmugam S, Boyer C. ACS Macro Lett. 2015; 4: 926
- 30 Shanmugam S, Xu J, Boyer C. Chem. Sci. 2015; 6: 1341
- 31 Zhou H, Johnson JA. Angew. Chem. Int. Ed. 2013; 52: 2235
- 32 Chen M, MacLeod MJ, Johnson JA. ACS Macro Lett. 2015; 4: 566
- 33 Goto A, Suzuki T, Ohfuji H, Tanishima M, Fukuda T, Tsujii Y, Kaji H. Macromolecules 2011; 44: 8709
- 34 Lei L, Tanishima M, Goto A, Kaji H. Polymers 2014; 6: 860
- 35 David G, Boyer C, Tonnar J, Ameduri B, Lacroix-Desmazes P, Boutevin B. Chem. Rev. 2006; 106: 3936
- 36 Ohtsuki A, Goto A, Kaji H. Macromolecules 2013; 46: 96
- 37 Ohtsuki A, Lei L, Tanishima M, Goto A, Kaji H. J. Am. Chem. Soc. 2015; 137: 5610
- 38 Barker IA, Dove AP. Chem. Commun. 2013; 49: 1205
- 39 Fu Q, McKenzie TG, Tan S, Nam E, Qiao GG. Polym. Chem. 2015; 6: 5362
- 40 Mol JC. J. Mol. Catal. A: Chem. 2004; 213: 39
- 41 Sutthasupa S, Shiotsuki M, Sanda F. Polym. J. 2010; 42: 905
- 42 Ogawa KA, Goetz AE, Boydston AJ. J. Am. Chem. Soc. 2015; 137: 1400
- 43 Goetz AE, Boydston AJ. J. Am. Chem. Soc. 2015; 137: 7572
Organic-based photoredox catalysts have also been utilized as photoinitiators, which is beyond the scope of this review. For examples, see:
-
References and Notes
- 1 Leibfarth FA, Mattson KM, Fors BP, Collins HA, Hawker CJ. Angew. Chem. Int. Ed. 2013; 52: 199
- 2 Yamago S, Nakamura Y. Polymer 2013; 54: 981
- 3 Fors BP, Hawker CJ. Angew. Chem. Int. Ed. 2012; 51: 8850
- 4 Mosnáček J, Ilčíková M. Macromolecules 2012; 45: 5859
- 5 Tasdelen MA, Uygun M, Yagci Y. Macromol. Chem. Phys. 2010; 211: 2271
- 6 Alfred NV, Jalapa NE, Morales SL, Ryabov AD, Le Lagadec R, Alexandrova L. Macromolecules 2012; 45: 8135
- 7 Telitel S, Dumur F, Campolo D, Poly J, Gigmes D, Fouassier JP, Lalevée J. J. Polym. Sci., Part A: Polym. Chem. 2015; DOI: 10.1002/pola.27896
- 8 Zhang T, Chen T, Amin I, Jordan R. Polym. Chem. 2014; 5: 4790
- 9 Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, de Alaniz JR, Fors BP, Hawker CJ. J. Am. Chem. Soc. 2014; 136: 16096
- 10 Pan X, Lamson M, Yan J, Matyjaszewski K. ACS Macro Lett. 2015; 4: 192
- 11 Matyjaszewski K, Pintauer T, Gaynor S. Macromolecules 2000; 33: 1476
- 12 Nicewicz DA, Nguyen TM. ACS Catal. 2014; 4: 355
- 13 Neumann M, Füldner S, König B, Zeitler K. Angew. Chem. Int. Ed. 2011; 50: 951
- 14 Hamilton DS, Nicewicz DA. J. Am. Chem. Soc. 2012; 134: 18577
- 15 Nguyen T, Nicewicz DA. J. Am. Chem. Soc. 2013; 135: 9588
- 16a Perkowski AJ, You W, Nicewicz DA. J. Am. Chem. Soc. 2015; 137: 7580
- 16b Xiao P, Zhang J, Dumur F, Tehfe MA, Morlet-Savary F, Graff B, Gigmes D, Fouassier JP, Lalevée J. Prog. Polym. Sci. 2015; 41: 32
- 17 Pintauer T, Matyjaszewski K. Chem. Soc. Rev. 2008; 37: 1087
- 18 Treat NJ, Fors BP, Kramer JW, Christianson M, Chiu CY, de Alaniz JR, Hawker CJ. ACS Macro Lett. 2014; 3: 580
- 19 Majek M, Filace F, von Wangelin AJ. Beilstein J. Org. Chem. 2014; 10: 981
- 20 Nguyen JD, D’Amato EM, Narayanam JM. R, Stephenson CR. J. Nat. Chem. 2012; 4: 854
- 21 Miyake GM, Theriot JC. Macromolecules 2014; 47: 8255
- 22 Moad G, Rizzardo E, Thang SH. Polymer 2008; 49: 1079
- 23 Xu J, Jung K, Atme A, Shanmugam S, Boyer C. J. Am. Chem. Soc. 2014; 136: 5508
- 24 Fu C, Xu J, Tao L, Boyer C. ACS Macro Lett. 2014; 3: 633
- 25 Xu J, Shanmugam S, Duong HT, Boyer C. Polym. Chem. 2015; 6: 5615
- 26 Quinn J, Barner L, Barner-Kowollik C, Rizzardo E, Davis TP. Macromolecules 2002; 35: 7620
- 27 Wang H, Li Q, Dai J, Du F, Zheng H, Bai R. Macromolecules 2013; 46: 2576
- 28 Goto A, Ohtsuki A, Ohfuji H, Tanishima M, Kaji H. J. Am. Chem. Soc. 2013; 135: 13286
- 29 Xu J, Shanmugam S, Boyer C. ACS Macro Lett. 2015; 4: 926
- 30 Shanmugam S, Xu J, Boyer C. Chem. Sci. 2015; 6: 1341
- 31 Zhou H, Johnson JA. Angew. Chem. Int. Ed. 2013; 52: 2235
- 32 Chen M, MacLeod MJ, Johnson JA. ACS Macro Lett. 2015; 4: 566
- 33 Goto A, Suzuki T, Ohfuji H, Tanishima M, Fukuda T, Tsujii Y, Kaji H. Macromolecules 2011; 44: 8709
- 34 Lei L, Tanishima M, Goto A, Kaji H. Polymers 2014; 6: 860
- 35 David G, Boyer C, Tonnar J, Ameduri B, Lacroix-Desmazes P, Boutevin B. Chem. Rev. 2006; 106: 3936
- 36 Ohtsuki A, Goto A, Kaji H. Macromolecules 2013; 46: 96
- 37 Ohtsuki A, Lei L, Tanishima M, Goto A, Kaji H. J. Am. Chem. Soc. 2015; 137: 5610
- 38 Barker IA, Dove AP. Chem. Commun. 2013; 49: 1205
- 39 Fu Q, McKenzie TG, Tan S, Nam E, Qiao GG. Polym. Chem. 2015; 6: 5362
- 40 Mol JC. J. Mol. Catal. A: Chem. 2004; 213: 39
- 41 Sutthasupa S, Shiotsuki M, Sanda F. Polym. J. 2010; 42: 905
- 42 Ogawa KA, Goetz AE, Boydston AJ. J. Am. Chem. Soc. 2015; 137: 1400
- 43 Goetz AE, Boydston AJ. J. Am. Chem. Soc. 2015; 137: 7572
Organic-based photoredox catalysts have also been utilized as photoinitiators, which is beyond the scope of this review. For examples, see: