

Subscribe to RSS
DOI: 10.1055/s-0037-1611744
Synthesis and Characterization of Acridinium Dyes for Photoredox Catalysis
Authors
This project was supported by Award No. R01 GM098340 from the National Institute of General Medical Sciences and a Camille Dreyfus Teacher-Scholar Award (D.A.N.). L.W. was supported by the International Postdoctoral Exchange Fellowship Program. Photophysical measurements were performed in the UNC-ERFC Instrumentation Facility established by the UNC-EFRC (Center for Solar Fuels, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001011).
Publication History
Received: 21 January 2019
Accepted after revision: 12 February 2019
Publication Date:
12 March 2019 (online)
Abstract
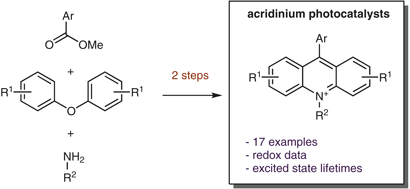
Photoredox catalysis is a rapidly evolving platform for synthetic methods development. The prominent use of acridinium salts as a sustainable option for photoredox catalysts has driven the development of more robust and synthetically useful versions based on this scaffold. However, more complicated syntheses, increased cost, and limited commercial availability have hindered the adoption of these catalysts by the greater synthetic community. By utilizing the direct conversion of a xanthylium salt into the corresponding acridinium as the key transformation, we present an efficient and scalable preparation of the most synthetically useful acridinium reported to date. This divergent strategy also enabled the preparation of a suite of novel acridinium dyes, allowing for a systematic investigation of substitution effects on their photophysical properties.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/s-0037-1611744.
- Supporting Information (PDF) (opens in new window)
-
References and Notes
- 1a Prier CK, Rankic DA, MacMillan DW. C. Chem. Rev. 2013; 113: 5322
- 1b Narayanam JM. R, Stephenson CR. J. Chem. Soc. Rev. 2011; 40: 102
- 1c Yoon TP, Ischay MA, Du J. Nat. Chem. 2010; 2: 527
- 1d Nicewicz DA, Nguyen TM. ACS Catal. 2014; 4: 355
- 1e Romero NA, Nicewicz DA. Chem. Rev. 2016; 116: 10075
- 1f Shaw MH, Twilton J, MacMillan DW. C. J. Org. Chem. 2016; 81: 6898
- 1g Wang C.-S, Dixneuf PH, Soulé J.-F. Chem. Rev. 2018; 118: 7532
- 2a Fukuzumi S, Kotani H, Ohkubo K, Ogo S, Tkachenko NV, Lemmetylnen H. J. Am. Chem. Soc. 2004; 126: 1600
- 2b Kotani H, Ohkubo K, Fukuzumi S. J. Am. Chem. Soc. 2004; 126: 15999
- 3 Joshi-Pangu A, Lévesque F, Roth HG, Oliver SF, Campeau L.-C, Nicewicz D, DiRocco DA. J. Org. Chem. 2016; 81: 7244
- 4 Acridinium 2 can be purchased for US $871/g from Sigma–Aldrich at the time of this publication.
- 5 Romero NA, Margrey KA, Tay NE, Nicewicz DA. Science 2015; 349: 1326
- 6a Fischer C, Sparr C. Angew. Chem. Int. Ed. 2018; 57: 2436
- 6b Fischer C, Sparr C. Tetrahedron 2018; 74: 5486
- 7a Katritzky AR, Brownlee RT. C, Musumarra G. Tetrahedron 1980; 36: 1643
- 7b Katritzky AR, Manzo RH, Lloyd JM, Patel RC. Angew. Chem. Int. Ed. Engl. 1980; 19: 306
- 8 Wu D, Feng X, Takase M, Haberecht MC, Müllen K. Tetrahedron 2008; 64: 11379
- 9 Birman VB, Chopra A, Ogle CA. Tetrahedron Lett. 1996; 37: 5073
- 10 Gram-Scale Preparation of Acridinium Salts; Typical Procedure for Acridinium 2 3,6-Di-tert-butyl-9-mesitylxanthylium Tetrafluoroborate (3) To a flame-dried 250 mL round-bottom flask under argon were added 4 (8.02 g, 28.4 mmol, 1 equiv), TMEDA (8.71 mL, 58.2 mmol, 2.05 equiv), and anhydrous n-hexane (28 mL). The resulting solution was cooled in an ice bath and sec-butyllithium (1.4 M solution in cyclohexane, 42.0 mL, 58.2 mmol, 2.05 equiv) was added dropwise. The ice bath was removed and the reaction mixture was stirred at room temperature for 4 h. The reaction was cooled to –78 °C and a solution of methyl 2,4,6-trimethylbenzoate (5.11 g, 28.7 mmol, 1.01 equiv) in anhydrous n-hexane (28 mL) was added slowly via cannula. After the addition, the mixture was allowed to slowly warm to room temperature and stirred for 12 h. The reaction was quenched with water (25 mL) and the biphasic mixture was stirred vigorously for 30 min. The mixture was diluted with Et2O (100 mL) and the layers were separated. The organic layer was washed with water (2 × 150 mL) and brine (1 × 150 mL). The organic layer was transferred to a 250 mL round-bottom flask equipped with a stir bar. To the vigorously stirred solution was added conc. HCl (12 mL), resulting in a bright-yellow precipitate. The suspension was stirred vigorously for 30 min then diluted with water (150 mL). The layers were separated and the organic layer was extracted with water (3 × 150 mL or until the washings become colorless). To the combined aqueous layers was added solid NaBF4 (9.35 g, 85.2 mmol, 3 equiv), resulting in a bright-yellow precipitate. The resulting suspension was extracted with dichloromethane (3 × 150 mL or until the washings become colorless). To the combined organic layers was added HBF4·Et2O complex (3.46 mL, 28.4 mmol, 1 equiv). The solution was swirled to achieve homogeneity then washed with water (1 × 100 mL) and aq. NaBF4 (1 M, 1 × 100 mL). The organic layer was dried over solid NaBF4, filtered, and concentrated to dryness. The residue was purified by trituration with hexanes and filtered. The solid was rinsed with n-pentane and dried in vacuo to give xanthylium 3 (10.6 g, 21.3 mmol, 75% yield) as a yellow-orange solid. 1H NMR (600 MHz, CDCl3): δ = 8.50 (s, 2 H), 7.97–7.88 (m, 2 H), 7.76 (d, J = 8.9 Hz, 2 H), 7.17 (s, 2 H), 2.49 (s, 3 H), 1.87 (s, 6 H), 1.54 (s, 18 H). 13C NMR (151 MHz, CDCl3): δ = 174.2, 171.0, 158.4, 141.3, 135.3, 129.2, 129.1, 128.5, 127.4, 122.0, 116.7, 37.5, 30.4, 21.3, 20.1. 19F NMR (376 MHz, CDCl3): δ = –153.90, –153.96. HRMS (ESI+): m/z [M]+ calcd for C30H35O: 411.2688; found: 411.2687. 3,6-Di-tert-butyl-9-mesityl-10-phenylacridin-10-ium Tetrafluoroborate (2): To an oven-dried 250 mL round-bottom flask under argon were added 3,6-di-tert-butyl-9-mesitylxanthylium tetrafluoroborate 3 (10.0 g, 20.1 mmol, 1 equiv) and dry, degassed dichloromethane (40 mL). To the resulting solution were added acetic acid (3.40 mL, 60.2 mmol, 3 equiv) followed by NEt3 (4.20 mL, 30.1 mmol, 1.5 equiv). Aniline (2.20 mL, 24.1 mmol, 1.2 equiv) was then added dropwise. The flask was covered with aluminum foil and stirred at room temperature for 12 h. The reaction was transferred to a separatory funnel and washed with water (1 × 50 mL) followed by sat. aq. NaHCO3 (1 × 50 mL). To the organic layer was added HBF4·Et2O complex (2.44 mL, 20.1 mmol, 1 equiv). The solution was swirled to achieve homogeneity then washed with water (1 × 100 mL) and aq. NaBF4 (1 M, 1 × 100 mL). The organic layer was dried over solid NaBF4, filtered, and concentrated to dryness. The residue was purified by trituration with 1:2 Et2O/hexanes and filtered. The solid was rinsed with n-pentane and dried in vacuo to give acridinium 2 (10.5 g, 18.3 mmol, 91% yield) as a bright-yellow solid. 1H NMR (500 MHz, CDCl3): δ = 7.96 (t, J = 7.6 Hz, 2 H), 7.90 (t, J = 7.5 Hz, 1 H), 7.83–7.75 (m, 4 H), 7.72 (d, J = 7.7 Hz, 2 H), 7.40 (s, 2 H), 7.16 (s, 2 H), 2.48 (s, 3 H), 1.85 (s, 6 H), 1.28 (s, 18 H). 13C NMR (151 MHz, CDCl3): δ = 163.6, 162.3, 142.1, 140.2, 136.8, 136.1, 131.8, 131.6, 129.2, 128.9, 128.3, 128.0, 127.5, 124.0, 115.0, 36.6, 30.2, 21.3, 20.2. 19F NMR (376 MHz, CDCl3): δ = –154.39, –154.44. HRMS (ESI+): m/z [M]+ calcd for C36H40N: 486.3161; found: 486.3157
- 11 Romero N, Nicewicz DA. J. Am. Chem. Soc. 2014; 136: 17024
- 12 Benniston AC, Elliott KJ, Harrington RW, Clegg W. Eur. J. Org. Chem. 2009; 253
- 13 As of February 2019, the 3-tert-butyl phenol required for the synthesis of biaryl ether 4 can be purchased for approximately US $160/mol (available in 500 g quantities from Frontier Scientific). For comparison, the 3-tert-butyl aniline required for the previous synthesis of 2 can be purchased for approximately US $707/mol (available in 100 g quantities from Combi-Blocks). Furthermore, the 2-bromo-4-(tert-butyl)benzoic acid coupling partner is prohibitively expensive (upwards of US $100,000/mol) and must be synthesized.
For recent reviews, see: