

Subscribe to RSS
DOI: 10.1055/a-1472-6852
Synthesis and Self-Assembly Behavior of Double Ullazine-Based Polycyclic Aromatic Hydrocarbons
Authors
Funding Information This research was financially supported by the EU Graphene Flagship (Graphene Core 3, 881603), ERC Consolidator Grant (T2DCP, 819698), the German Research Foundation (DFG) within the Cluster of Excellence “Center for Advancing Electronics Dresden (cfaed)” and DFG-NSFC Joint Sino-German Research Project (EnhanceNano, No. 391979941), as well as the DFG-SNSF Joint Switzerland-German Research Project (EnhanTopo, No. 429265950). M. Borkowski and T. Marszalek acknowledge the Foundation for Polish Science financed by the European Union under the European Regional Development Fund (POIR.04.04.00-00-3ED8/17). W. Pisula acknowledges National Science Centre, Poland, through the grant UMO-2015/18/E/ST3/00322.

Abstract
Polycyclic aromatic azomethine ylides (PAMY, 1) are versatile building blocks for the bottom-up synthesis of nitrogen-containing polycyclic aromatic hydrocarbons (N-PAHs). Although the chemistry of PAMY was already established few years ago, the cycloaddition of a double PAMY building block has not been reported so far. In this work, we demonstrate the first cycloaddition of a PAMY-dimer (6), which opens the access to three different alkyl ester-substituted N-PAHs with a laterally extended double ullazine scaffold (DU-1, DU-2 and DU-3). Interestingly, the cyclic voltammetry of DU-1–3 exhibited three reversible oxidation waves, which confirmed the electron-rich nature of the double ullazine scaffold. Furthermore, in situ spectroelectrochemistry study of ethylhexyl ester-substituted DU-3 revealed the formation of different cationic species with new absorption bands up to 1689 nm. Additionally, the influence of the attached substituents on the film formation and supramolecular organization in the thin films was investigated by polarized optical microscopy and grazing incidence wide-angle X-ray scattering.
Key words
polycyclic aromatic hydrocarbons - nanographenes - polycyclic aromatic azomethine ylides - cycloaddition - ullazineIntroduction
Polycyclic aromatic hydrocarbons (PAHs), which can be considered as small cutouts of graphene, are a remarkable class of organic compounds with unique (opto)electronic properties.[1] A continuous effort in the advancement in the reaction methodology and molecular design for the development of functionalized PAHs was devoted in order to tune their (opto)electronic properties and to enable an implementation into organic electronics.[2] One efficient pathway to tailor the intrinsic optical and electronic properties of PAHs is the introduction of nitrogen atoms, which may lead to control over the energy level of the frontier orbital or the stabilization of charges and radicals.[3] For example, the nitrogen-containing PAHs (N-PAHs) with a 16 π-electron ullazine motif have evolved as an attractive class of PAHs for dye-sensitized solar cells.[4] Although the first ullazine derivatives were already reported by Zeller in 1983,[5] the synthesis of ullazine-embedded PAHs still remains challenging and is mostly limited to acid-promoted and metal-catalyzed cyclization reactions.[6]
In 2014, our group firstly reported the synthesis of polycyclic aromatic azomethine ylides (PAMYs, 1), which are unique building blocks for the construction of unprecedented N-PAHs via a radical or zwitterionic pathway ([Figure 1a]).[7] Especially, the 1,3-dipolar cycloaddition between PAMY (1) and alkenes/alkynes (2) with subsequent dehydrogenation is a powerful tool for the formation of ullazine-based PAHs (3).[8] Moreover, the 1,3-dipolar cycloaddition with PAMY (1) was recently extended to the use of nitriles (4) as dipolarophiles and allowed the formation of azaullazine derivatives (5) on surface as well as in solution.[9] While the cycloaddition of the PAMY building block (1) has already been intensively investigated in the last few years, the 1,3-dipolar cycloaddition of an extended PAMY dimer is not known so far.

In this work, we demonstrate the first cycloaddition of PAMY-dimer (6), which allows the synthesis of three novel alkyl ester-substituted N-PAHs with a laterally extended double ullazine scaffold (DU-1, DU-2 and DU-3, [Figure 1b]). The optoelectronic properties of DU-1–3 are comprehensively investigated by UV-Vis absorption spectroscopy, fluorescence spectroscopy and cyclic voltammetry (CV) as well as supported by theoretical modelling via density functional theory (DFT) calculations. In particular, the CV of DU-1–3 showed three reversible oxidations waves, which confirmed the electron-rich structure of the double-ullazine framework. Moreover, spectroelectrochemistry (SEC) measurements unraveled several cationic species for DU-3, which were verified by UV-Vis-NIR absorption and electron paramagnetic resonance (EPR) spectroscopies. Furthermore, the self-organization of DU-1–3 was investigated by polarized optical microscopy (POM) and grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements. In contrast to linear alkyl ester-substituted DU-1 and DU-2 (C10 to C12 ), the assemblies of the branched alkyl ester-substituted DU-3 (C 7,2) showed the highest crystallinity.
Results and Discussion
The target compounds DU-1–3 were synthesized starting from the tetra-alcohol species 7 ([Scheme 1]). The synthesis of 7 was carried out according to our previous synthetic route.[10] The HCl-induced microwave-assisted cyclization of 7 and subsequent hydride abstraction with tritylium tetrafluoroborate gave the iminium salt 8 as a crude product.[11] Afterwards, the addition of triethylamine (TEA) to the crude iminium salt and corresponding dipolarophiles (9, 10 or 11) at 60 °C enabled the twofold cycloaddition. The following oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) provided the target compounds DU-1–3, respectively, as yellow solids in yields from 58% for DU-1 up to 59% for DU-3 and 61% for DU-2, over two steps.[12] All target compounds DU-1–3 were confirmed by NMR spectroscopy in C2D2Cl4 (see the Supporting Information, SI). The aromatic protons of DU-1–3 in the 1H-NMR spectrum are assigned to the expected chemical structure by the assistance of two-dimensional NMR spectroscopy, respectively (see SI). Furthermore, DU-1–3 were characterized by high-resolution (HR) matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectroscopy (MS). In detail, the MALDI-TOF isotopic distributions of DU-1–3 are in perfect agreement with the simulated patterns (see SI). Due to the different alkyl ester substitutions, the obtained DU-1–3 showed a different solubility behavior in common organic solvents, such as dichloromethane (DCM). DU-1 and DU-2 with linear C10- and C12-ester substitutions, respectively, provided a low solubility in DCM (0.2 mg/mL). In contrast, DU-3 with branched C7,2 ester substitutions showed an enhanced solubility of up to 3 mg/mL in DCM. Differential scanning calorimetry reveals only one peak for all three compounds that is related to the phase transition from the crystalline to the isotropic phase (SI, [Figure S5]). The phase transition temperature decreases with longer and more space-demanding side chains from 222.4 °C for DU-1 to 206°C for DU-2 and 203.8 °C for DU-3. Thermogravimetric analysis reveals a high thermal stability of DU-1–3 up to 260 °C (SI, [Figure S6]).

The optoelectronic properties of DU-1–3 were investigated by UV-Vis absorption and fluorescence spectroscopy in anhydrous DCM. From the UV-Vis absorption and fluorescence spectra, there were no differences between DU-1, DU-2 and DU-3, suggesting that the different alkyl ester-substituents do not apparently influence the optoelectronic properties (see SI). Due to the similarity of the optoelectronic properties, only the branched ethylhexyl ester-substituted DU-3 are exemplarily discussed here. The absorption maximum (λabs) for DU-3 was observed at 404 nm with two shoulder peaks at 427 and 446 nm (see [Figure 2a]). The corresponding optical energy gap (ΔE g) was estimated from the onset of the UV-Vis absorption spectrum and was calculated to be 2.68 eV for DU-3. The time-dependent (TD) DFT calculations at the B3LYP/6-31G(d) level fitted the experimental UV-Vis absorption result. The fluorescence spectrum of dimer DU-3 revealed a fluorescence maximum at 461 nm with a shoulder at 494 nm ([Figure 2b]). The fluorescence quantum yields varied in the range from 32.9% for DU-2 up to 37.6% and 37.7% for DU-1 and for DU-3, respectively.
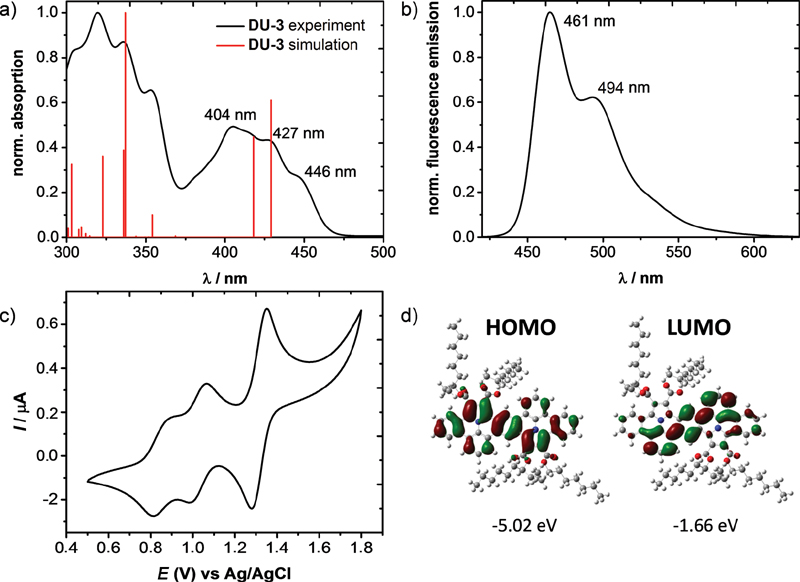
The electrochemical properties of DU-1–3 were investigated by CV measurements in anhydrous DCM. Due to the different solubility in DCM, the intensities of the CV measurements of DU-1–3 are diverged (see SI, [Figure S4]). Due to the similarity, the CV data of DU-3 with the best solubility in DCM are presented in [Figure 2c]. Compound DU-3 offered reversible two oxidation waves at 0.3 and 0.5 V versus Fc/Fc+. In comparison to the first two oxidation waves, the third oxidation peak at 0.78 V has a much higher intensity, which presumably indicates the overlap of two closely spaced oxidation processes or a two-electron oxidation. Nevertheless, a reduction behavior was not observed in the available potential window. The corresponding HOMO level of DU-3 was estimated to be −5.1 eV by the half-wave potential of the first reversible oxidation. The LUMO of DU-3, which was determined from the difference between the optical energy gap and electrochemical HOMO, is −2.42 eV. DFT and TD-DFT calculations with a Gaussian 09 package were performed for a deeper grasp of the electronic ground state. The geometry optimization was carried out via the B3LYP level of theory with the 6-31G(d) basis set. The graphical representations of the HOMO and LUMO of DU-3 are provided in [Figure 2d] (DU-1 and DU-2: see SI, [Figure S11]). The HOMO of DU-3 is completely symmetric and equally delocalized over the full π-system. The DFT-calculated HOMO levels for DU-1–3 are in perfect agreement with the experimental HOMO levels derived from the CV measurements (SI, [Table S5]). In contrast, the distribution of the LUMO shows the main localization between the two ullazine units.
Due to the highly reversible oxidation behavior of DU-3, detailed insight into the cationic species was achieved by SEC via in situ CV, UV-Vis-NIR absorption and EPR spectroscopy in anhydrous DCM (see [Figure 3]). At low positive potentials, new absorption bands at 493, 570, 1380 and 1689 nm are associated to the formation of the radical cation species ([Figure 3a]). The appearance of the EPR signal confirmed the formation of the radical cation DU-3•+ . The EPR spectrum of the radical showed a broad unresolved signal with a g-value of 2.0027 ([Figure 3c]). The DFT calculations demonstrated that the spin density of DU-3•+ is delocalized over two ullazine motifs in agreement with the shape of the HOMO ([Figure 3d]). During the second redox process, the intensities of bands centered at 493 and 570 nm increased further and the bands peaked at 1380 and 1689 nm are blue-shifted. The EPR signal intensity showed a two-fold increase during the second oxidation process ([Figure 3b]), indicating that the dication DU-32+ may have a diradical character. At the potentials of the third redox event in the CV, the EPR signal intensity decreased and new absorption bands emerged at 531 and 834 nm. This is an indication for the formation of the EPR-silent four positively charged species DU-34+ .

[Figure 4(a–c)] shows the POM images of DU-1–3 drop-cast films. All layers exhibit birefringence between cross-polarizers and the light intensity of the entire crystals changes from bright to dark by 45° rotation of the substrate. DU-1 and DU-2 form a comparable surface morphology with spherulitic domains that are less than 100 micrometers in size. In contrast to DU-1-2, DU-3 forms much larger polycrystalline structures with diameters larger than 100 micrometers. This suggests that the branched substituents improve the film crystallinity and molecular order.

To understand the supramolecular organization of the DUs, GIWAXS of the polycrystalline drop-cast film was performed ([Figure 4d–f]). The GIWAXS patterns reveal significant differences of the three compounds in crystallinity. All compounds exhibited certain edge-on arrangement on the substrate as indicated by the maximum intensities of the out-of-plane h00 (according to the Miller index) and in-plane 00l interstack, as well as π-stacking reflections (0k0). The out-of-plane interstack distance is closely related to the length of the substituents and decreased from 2.82 nm for DU-2 to 2.61 nm for DU-1, and 2.20 nm for DU-3. This correlation suggests that the long axis of the PAHs is oriented parallel to the surface. DU-1 and DU-2 reveal a close π-stacking distance of 0.35 nm, but both are poorly ordered in the in-plane as indicated by a low intensity of the corresponding reflection. Although the molecular interactions might be reduced due to the steric hindrance of the branched substituents,[13] the π-stacking distance of 0.35 nm for DU-3 remains unchanged. The further equatorial reflection located at qxy = 0.47 Å−1 and qz = 0 Å−1 ([Figure 4d]) is assigned to the in-plane 00l interstack distance of 1.33 nm for DU-1 and DU-2, and 1.07 nm for DU-3. Since the theoretical molecular length of 1.62 nm calculated by Cerius[2] software is larger, it is assumed that the molecules are arranged in a herringbone structure. As already observed by POM, the crystallinity of DU-3 is improved in comparison to DU-1-2 as evidenced by the additional high-intensity reflections. In summary of the structural study, it can be concluded that the introduction of branched side chains in DU-3 enhances the self-assembly and crystallinity.
Conclusions
In summary, we have synthesized a series of alkyl ester-substituted N-PAHs with a laterally extended double ullazine scaffold (DU-1–3). As key step for the synthesis, the first example of the double cycloaddition between PAMY-dimer (6) and different electron-deficient dipolarophiles was presented. Interestingly, the CV measurements for DU-1–3 revealed a highly reversible oxidation potential and confirmed up to three oxidation waves. The corresponding different oxidized species of DU-3 were investigated by in situ EPR measurements and UV-Vis-NIR spectroscopy. It was shown that the radical species with the pronounced absorption bands in the NIR region are formed. Additionally, the self-assembly of the different alkyl ester-substituted DU-1–3 was investigated by POM and GIWAXS. In contrast to linear alkyl ester-substituted derivatives (DU-1 and DU-2), the branched ethylhexyl ester-substituted compound (DU-3) revealed a higher order of the self-assembled structure. We believe that the cycloaddition of the double-ullazine based PAHs could pave the way for the synthesis of unprecedented extended N-PAHs or N-doped graphene nanoribbons.
No conflict of interest has been declared by the author(s).
Acknowledgment
We thank F. Drescher, Dr. S. Machill, Prof. E. Brunner and Dr. T. Lübken for HR-MS measurements and NMR measurements, respectively. We thank the Center for Information Services and High Performance Computing (ZIH) at TU Dresden for generous allocations of compute resources. Moreover, we thank M. Sc. Felix Fries and Prof. Sebastian Reineke for PLQY measurements.
Supporting Information
Supporting Information for this article is available online at https://doi.org/10.1055/a-1472-6852.
Dedicated to Professor Peter Bäuerle on the occasion of his 65th birthday.
-
References and Notes
- 1a Narita A, Wang X.-Y, Feng X, Müllen K. Chem. Soc. Rev. 2015; 44: 6616
- 1b Liu J, Feng X. Angew. Chem. Int. Ed. 2020; 59: 23386
- 2 Wang X.-Y, Narita A, Müllen K. Nat. Rev. Chem. 2017; 2: 0100
- 3a Liu J, Feng X. Synlett 2020; 31: 211
- 3b Ji L, Friedrich A, Krummenacher I, Eichhorn A, Braunschweig H, Moos M, Hahn S, Geyer FL, Tverskoy O, Han J, Lambert C, Dreuw A, Marder TB, Bunz UH. F. J. Am. Chem. Soc. 2017; 139: 15968
- 3c Takase M, Enkelmann V, Sebastiani D, Baumgarten M, Müllen K. Angew. Chem. Int. Ed. 2007; 46: 5524
- 4a Delcamp JH, Yella A, Holcombe TW, Nazeeruddin MK, Grätzel M. Angew. Chem. Int. Ed. 2013; 52: 376
- 4b Qiao H, Deng Y, Peng R, Wang G, Yuan J, Tan S. RSC Adv. 2016; 6: 70046
- 4c Mathew S, Astani NA, Curchod BF. E, Delcamp JH, Marszalek M, Frey J, Rothlisberger U, Nazeeruddin MK, Grätzel M. J. Mater. Chem. A 2016; 4: 2332
- 4d Cebrián C. J. Mater. Chem. C 2018; 6: 11943
- 5 Balli H, Zeller M. Helv. Chim. Acta 1983; 66: 2135
- 6a Zhang G, Gautam P, Chan JM. W. Org. Chem. Front. 2020; 7: 787
- 6b Das A, Ghosh I, König B. Chem. Commun. 2016; 52: 8695
- 6c Wan D, Li X, Jiang R, Feng B, Lan J, Wang R, You J. Org. Lett. 2016; 18: 2876
- 6d Boldt S, Parpart S, Villinger A, Ehlers P, Langer P. Angew. Chem. Int. Ed. 2017; 56: 4575
- 7a Berger R, Wagner M, Feng X, Müllen K. Chem. Sci. 2015; 6: 436
- 7b Wang X.-Y, Richter M, He Y, Björk J, Riss A, Rajesh R, Garnica M, Hennersdorf F, Weigand JJ, Narita A, Berger R, Feng X, Auwärter W, Barth JV, Palma C.-A, Müllen K. Nat. Commun. 2017; 8: 1948
- 8a Richter M, Schellhammer KS, Machata P, Cuniberti G, Popov A, Ortmann F, Berger R, Müllen K, Feng X. Org. Chem. Front. 2017; 4: 847
- 8b Richter M, Hahn S, Dmitrieva E, Rominger F, Popov A, Bunz UH. F, Feng X, Berger R. Chemistry 2019; 25: 1345
- 8c Ito S, Tokimaru Y, Nozaki K. Angew. Chem. 2015; 127: 7364
- 9a Riss A, Richter M, Paz AP, Wang X.-Y, Raju R, He Y, Ducke J, Corral E, Wuttke M, Seufert K, Garnica M, Rubio A, V Barth, J. Narita A, Müllen K, Berger R, Feng X, Palma C.-A, Auwärter W. Nat. Commun. 2020; 11: 1490
- 9b Li Q.-Q, Ochiai K, Lee C.-A, Ito S. Org. Lett. 2020; 22: 6132
- 10a Berger R, Giannakopoulos A, Ravat P, Wagner M, Beljonne D, Feng X, Müllen K. Angew. Chem. Int. Ed. 2014; 53: 10520
- 10b Berger R, Giannakopoulos A, Ravat P, Wagner M, Beljonne D, Feng X, Müllen K. Angew. Chem. 2014; 126: 10688
- 11
Synthetic procedure for iminium salt (8): In a microwave tube, the tetra-alcohol species (7, 0.1 g, 188 µmol) was added into a stirring anhydrous hydrogen chloride solution
(4 M in dioxane, 5 mL). The microwave tube was capped and placed in a microwave reactor.
A dynamic mode was chosen (300 W, power max: on, activated cooling, pre-stirring:
10 s, temperature: 130 °C) for 1.5 h. After cooling to room temperature, the cap was
removed and the reaction mixture was transferred to a round-bottom flask. The solvents
were removed under reduced pressure. The crude product was dissolved in toluene (anhydrous,
20 mL) and heated to 90 °C under argon. In a second dry and inert Schlenk flask, triphenylcarbenium
tetrafluoroborate was dissolved in anhydrous acetonitrile and added dropwise. After
continuous stirring for 2 h, the solvents were removed under reduced pressure. The
residue was dissolved in DCM and precipitated in diethyl ether (250 mL). The crude
product 8 was obtained as a yellow solid. HR-MS (MALDI-ToF):m/z ([M + H]+ ) = 459.1893, calcd. for C34H23N2: m/z = 459.1861, error = 6.96 ppm
- 12
General synthetic procedure of DU-1–3: In a dry and inert Schlenk flask, crude 8 (100 mg) and the corresponding dipolarophiles were dissolved in anhydrous chloroform.
At 60 °C, the addition of triethylamine was carried out in one shot and the reaction
mixture was kept under continuous stirring overnight. After cooling to room temperature,
the reaction mixture was transferred into a round-bottom flask and the solvent was
removed under reduced pressure. After the addition of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
(DDQ) the round-bottom flask was sealed and purged with argon. Anhydrous toluene was
added and the reaction mixture was stirred for 3 h. The reaction was quenched with
water and extracted with dichloromethane (50 mL, 5 times). The solvent was removed
under reduced pressure. The crude product was dissolved in a small amount of dichloromethane
and precipitated in methanol (250 mL). After the filtration, the crude product was
purified by column chromatography on silica in pure chloroform and via rGPC. The target
compounds DU-1–3 were obtained as yellow solids. DU-1:1H-NMR (600 MHz, C2D2Cl4): δ 9.71 (s, 2 H), 8.25 (d, J = 8.2 Hz, 2 H), 8.19 (d, J = 8.0 Hz, 2 H), 8.01 (d, J = 8.0 Hz, 2 H), 7.89 (d, J = 8.0 Hz, 2 H), 7.58 (t, J = 7.7 Hz, 2 H), 7.32 (t, J = 7.5 Hz, 2 H), 7.23 (t, J = 7.3 Hz, 2 H), 4.49 (t, J = 7.0 Hz, 4 H), 4.42 (t, J = 7.0 Hz, 4 H), 1.83 (td, J = 14.5, 7.2 Hz, 12 H), 1.53–1.40 (m, 8 H), 1.38–1.30 (m, 8 H), 1.29–1.25 (m, 4 H),
1.24–1.12 (m, 40 H), 0.77 (dt, J = 21.0, 7.0 Hz, 12 H). 13C-NMR (151 MHz, C2D2Cl4): δ 167.2, 166.2, 128.6, 128.0, 127.9 127.6, 125.7, 125.6, 124.6, 123.8, 123.0, 122.1,
122.0, 121.4, 32.2, 30.0, 29.9, 29.8, 29.7, 29.2, 29.0, 26.5, 23.0, 14.5. HR-MS (MALDI-ToF):
m/z ([M]+ ) = 1240.7471, calcd. for C82H100N2O8: m/z = 1240.7479, error = −0.7 ppm. IR: ṽ = 2921, 2853, 1713, 1195, 1128, 747 cm−1. DU-2:1H-NMR (600 MHz, C2D2Cl4): 9.54 (s, 2 H), 8.10 (d, J = 7.5 Hz, 2 H), 8.04 (d, J = 7.6 Hz, 2 H), 7.85 (d, J = 7.3 Hz, 2 H), 7.72 (d, J = 7.2 Hz, 2 H), 7.47 (t, J = 7.2 Hz, 2 H), 7.21 (t, J = 7.0 Hz, 2 H), 7.10 (t, J = 6.5 Hz, 2 H), 4.48–4.40 (m, 7 H), 4.40–4.27 (m, 8 H), 1.79 (d, J = 6.8 Hz, 8 H), 1.42 (d, J = 6.1 Hz, 8 H), 1.37–1.25 (m, 8 H), 1.12 (dd, J = 28.9, 25.8 Hz, 60 H), 0.72 (dd, J = 16.3, 6.8 Hz, 12 H). 13C-NMR (151 MHz, C2D2Cl4): δ 167.54, 166.39, 128.55, 127.94, 127.87, 126.50, 126.22, 125.64, 124.50, 124.07,
123.80, 123.42, 122.26, 122.20, 122.03, 121.32, 120.57, 120.07, 74.20, 32.20, 30.02,
30.00, 29.98, 29.93, 29.80, 29.67, 29.16, 29.03, 26.55, 26.52, 23.00, 14.51, 14.50.
HR-MS (MALDI-ToF): m/z ([M]+ ) = 1352.8741, calcd. for C90H116N2O8: m/z = 1352.8731, error = 0.7 ppm. IR: ṽ = 2918, 2851, 1713, 1194, 1126, 747 cm−1. DU-3:1H-NMR (600 MHz, C2D2Cl4): δ 9.78 (s, 2 H), 8.39 (dd, J = 24.5, 6.4 Hz, 4 H), 8.24 (d, J = 6.7 Hz, 2 H), 8.16 (s, 2 H), 7.69 (t, J = 7.5 Hz, 2 H), 7.42 (s, 4 H), 5.28–5.21 (m, 2 H), 5.17–5.04 (m, 2 H), 1.86–1.61
(m, 20 H), 1.34 (d, J = 37.1 Hz, 12 H), 1.26–1.14 (m, 12 H), 1.08 (d, J = 2.8 Hz, 8 H), 1.01–0.89 (m, 12 H), 0.81 (d, J = 1.1 Hz, 6 H), 0.66 (d, J = 6.2 Hz, 6 H). 13C-NMR (151 MHz, C2D2Cl4): δ 166.84, 166.79, 128.82, 128.68, 128.17, 127.11, 126.64, 126.62, 125.73, 125.72,
125.26, 125.24, 125.22, 124.67, 124.24, 124.14, 123.78, 123.27, 123.25, 122.87, 122.31,
122.29, 121.77, 120.13, 120.10, 116.85, 114.87, 114.38, 114.35, 74.20, 33.56, 33.46,
32.91, 32.11, 32.00, 29.80, 29.67, 26.83, 26.17, 25.72, 25.51, 22.98, 22.89, 14.51,
14.37, 10.06, 9.82. HR-MS (MALDI-ToF): m/z ([M]+ ) = 1184.6855, calcd. for C78H92N2O8: m/z = 1184.6853, error = 0.1 ppm. IR: ṽ = 2954, 2924, 2858, 1703, 1194, 744 cm−1
- 13 Osaka I, Zhang R, Sauvé G, Smilgies D.-M, Kowalewski T, McCullough RD. J. Am. Chem. Soc. 2009; 131: 2521
Publication History
Received: 26 January 2021
Accepted: 18 March 2021
Accepted Manuscript online:
01 April 2021
Article published online:
27 May 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References and Notes
- 1a Narita A, Wang X.-Y, Feng X, Müllen K. Chem. Soc. Rev. 2015; 44: 6616
- 1b Liu J, Feng X. Angew. Chem. Int. Ed. 2020; 59: 23386
- 2 Wang X.-Y, Narita A, Müllen K. Nat. Rev. Chem. 2017; 2: 0100
- 3a Liu J, Feng X. Synlett 2020; 31: 211
- 3b Ji L, Friedrich A, Krummenacher I, Eichhorn A, Braunschweig H, Moos M, Hahn S, Geyer FL, Tverskoy O, Han J, Lambert C, Dreuw A, Marder TB, Bunz UH. F. J. Am. Chem. Soc. 2017; 139: 15968
- 3c Takase M, Enkelmann V, Sebastiani D, Baumgarten M, Müllen K. Angew. Chem. Int. Ed. 2007; 46: 5524
- 4a Delcamp JH, Yella A, Holcombe TW, Nazeeruddin MK, Grätzel M. Angew. Chem. Int. Ed. 2013; 52: 376
- 4b Qiao H, Deng Y, Peng R, Wang G, Yuan J, Tan S. RSC Adv. 2016; 6: 70046
- 4c Mathew S, Astani NA, Curchod BF. E, Delcamp JH, Marszalek M, Frey J, Rothlisberger U, Nazeeruddin MK, Grätzel M. J. Mater. Chem. A 2016; 4: 2332
- 4d Cebrián C. J. Mater. Chem. C 2018; 6: 11943
- 5 Balli H, Zeller M. Helv. Chim. Acta 1983; 66: 2135
- 6a Zhang G, Gautam P, Chan JM. W. Org. Chem. Front. 2020; 7: 787
- 6b Das A, Ghosh I, König B. Chem. Commun. 2016; 52: 8695
- 6c Wan D, Li X, Jiang R, Feng B, Lan J, Wang R, You J. Org. Lett. 2016; 18: 2876
- 6d Boldt S, Parpart S, Villinger A, Ehlers P, Langer P. Angew. Chem. Int. Ed. 2017; 56: 4575
- 7a Berger R, Wagner M, Feng X, Müllen K. Chem. Sci. 2015; 6: 436
- 7b Wang X.-Y, Richter M, He Y, Björk J, Riss A, Rajesh R, Garnica M, Hennersdorf F, Weigand JJ, Narita A, Berger R, Feng X, Auwärter W, Barth JV, Palma C.-A, Müllen K. Nat. Commun. 2017; 8: 1948
- 8a Richter M, Schellhammer KS, Machata P, Cuniberti G, Popov A, Ortmann F, Berger R, Müllen K, Feng X. Org. Chem. Front. 2017; 4: 847
- 8b Richter M, Hahn S, Dmitrieva E, Rominger F, Popov A, Bunz UH. F, Feng X, Berger R. Chemistry 2019; 25: 1345
- 8c Ito S, Tokimaru Y, Nozaki K. Angew. Chem. 2015; 127: 7364
- 9a Riss A, Richter M, Paz AP, Wang X.-Y, Raju R, He Y, Ducke J, Corral E, Wuttke M, Seufert K, Garnica M, Rubio A, V Barth, J. Narita A, Müllen K, Berger R, Feng X, Palma C.-A, Auwärter W. Nat. Commun. 2020; 11: 1490
- 9b Li Q.-Q, Ochiai K, Lee C.-A, Ito S. Org. Lett. 2020; 22: 6132
- 10a Berger R, Giannakopoulos A, Ravat P, Wagner M, Beljonne D, Feng X, Müllen K. Angew. Chem. Int. Ed. 2014; 53: 10520
- 10b Berger R, Giannakopoulos A, Ravat P, Wagner M, Beljonne D, Feng X, Müllen K. Angew. Chem. 2014; 126: 10688
- 11
Synthetic procedure for iminium salt (8): In a microwave tube, the tetra-alcohol species (7, 0.1 g, 188 µmol) was added into a stirring anhydrous hydrogen chloride solution
(4 M in dioxane, 5 mL). The microwave tube was capped and placed in a microwave reactor.
A dynamic mode was chosen (300 W, power max: on, activated cooling, pre-stirring:
10 s, temperature: 130 °C) for 1.5 h. After cooling to room temperature, the cap was
removed and the reaction mixture was transferred to a round-bottom flask. The solvents
were removed under reduced pressure. The crude product was dissolved in toluene (anhydrous,
20 mL) and heated to 90 °C under argon. In a second dry and inert Schlenk flask, triphenylcarbenium
tetrafluoroborate was dissolved in anhydrous acetonitrile and added dropwise. After
continuous stirring for 2 h, the solvents were removed under reduced pressure. The
residue was dissolved in DCM and precipitated in diethyl ether (250 mL). The crude
product 8 was obtained as a yellow solid. HR-MS (MALDI-ToF):m/z ([M + H]+ ) = 459.1893, calcd. for C34H23N2: m/z = 459.1861, error = 6.96 ppm
- 12
General synthetic procedure of DU-1–3: In a dry and inert Schlenk flask, crude 8 (100 mg) and the corresponding dipolarophiles were dissolved in anhydrous chloroform.
At 60 °C, the addition of triethylamine was carried out in one shot and the reaction
mixture was kept under continuous stirring overnight. After cooling to room temperature,
the reaction mixture was transferred into a round-bottom flask and the solvent was
removed under reduced pressure. After the addition of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
(DDQ) the round-bottom flask was sealed and purged with argon. Anhydrous toluene was
added and the reaction mixture was stirred for 3 h. The reaction was quenched with
water and extracted with dichloromethane (50 mL, 5 times). The solvent was removed
under reduced pressure. The crude product was dissolved in a small amount of dichloromethane
and precipitated in methanol (250 mL). After the filtration, the crude product was
purified by column chromatography on silica in pure chloroform and via rGPC. The target
compounds DU-1–3 were obtained as yellow solids. DU-1:1H-NMR (600 MHz, C2D2Cl4): δ 9.71 (s, 2 H), 8.25 (d, J = 8.2 Hz, 2 H), 8.19 (d, J = 8.0 Hz, 2 H), 8.01 (d, J = 8.0 Hz, 2 H), 7.89 (d, J = 8.0 Hz, 2 H), 7.58 (t, J = 7.7 Hz, 2 H), 7.32 (t, J = 7.5 Hz, 2 H), 7.23 (t, J = 7.3 Hz, 2 H), 4.49 (t, J = 7.0 Hz, 4 H), 4.42 (t, J = 7.0 Hz, 4 H), 1.83 (td, J = 14.5, 7.2 Hz, 12 H), 1.53–1.40 (m, 8 H), 1.38–1.30 (m, 8 H), 1.29–1.25 (m, 4 H),
1.24–1.12 (m, 40 H), 0.77 (dt, J = 21.0, 7.0 Hz, 12 H). 13C-NMR (151 MHz, C2D2Cl4): δ 167.2, 166.2, 128.6, 128.0, 127.9 127.6, 125.7, 125.6, 124.6, 123.8, 123.0, 122.1,
122.0, 121.4, 32.2, 30.0, 29.9, 29.8, 29.7, 29.2, 29.0, 26.5, 23.0, 14.5. HR-MS (MALDI-ToF):
m/z ([M]+ ) = 1240.7471, calcd. for C82H100N2O8: m/z = 1240.7479, error = −0.7 ppm. IR: ṽ = 2921, 2853, 1713, 1195, 1128, 747 cm−1. DU-2:1H-NMR (600 MHz, C2D2Cl4): 9.54 (s, 2 H), 8.10 (d, J = 7.5 Hz, 2 H), 8.04 (d, J = 7.6 Hz, 2 H), 7.85 (d, J = 7.3 Hz, 2 H), 7.72 (d, J = 7.2 Hz, 2 H), 7.47 (t, J = 7.2 Hz, 2 H), 7.21 (t, J = 7.0 Hz, 2 H), 7.10 (t, J = 6.5 Hz, 2 H), 4.48–4.40 (m, 7 H), 4.40–4.27 (m, 8 H), 1.79 (d, J = 6.8 Hz, 8 H), 1.42 (d, J = 6.1 Hz, 8 H), 1.37–1.25 (m, 8 H), 1.12 (dd, J = 28.9, 25.8 Hz, 60 H), 0.72 (dd, J = 16.3, 6.8 Hz, 12 H). 13C-NMR (151 MHz, C2D2Cl4): δ 167.54, 166.39, 128.55, 127.94, 127.87, 126.50, 126.22, 125.64, 124.50, 124.07,
123.80, 123.42, 122.26, 122.20, 122.03, 121.32, 120.57, 120.07, 74.20, 32.20, 30.02,
30.00, 29.98, 29.93, 29.80, 29.67, 29.16, 29.03, 26.55, 26.52, 23.00, 14.51, 14.50.
HR-MS (MALDI-ToF): m/z ([M]+ ) = 1352.8741, calcd. for C90H116N2O8: m/z = 1352.8731, error = 0.7 ppm. IR: ṽ = 2918, 2851, 1713, 1194, 1126, 747 cm−1. DU-3:1H-NMR (600 MHz, C2D2Cl4): δ 9.78 (s, 2 H), 8.39 (dd, J = 24.5, 6.4 Hz, 4 H), 8.24 (d, J = 6.7 Hz, 2 H), 8.16 (s, 2 H), 7.69 (t, J = 7.5 Hz, 2 H), 7.42 (s, 4 H), 5.28–5.21 (m, 2 H), 5.17–5.04 (m, 2 H), 1.86–1.61
(m, 20 H), 1.34 (d, J = 37.1 Hz, 12 H), 1.26–1.14 (m, 12 H), 1.08 (d, J = 2.8 Hz, 8 H), 1.01–0.89 (m, 12 H), 0.81 (d, J = 1.1 Hz, 6 H), 0.66 (d, J = 6.2 Hz, 6 H). 13C-NMR (151 MHz, C2D2Cl4): δ 166.84, 166.79, 128.82, 128.68, 128.17, 127.11, 126.64, 126.62, 125.73, 125.72,
125.26, 125.24, 125.22, 124.67, 124.24, 124.14, 123.78, 123.27, 123.25, 122.87, 122.31,
122.29, 121.77, 120.13, 120.10, 116.85, 114.87, 114.38, 114.35, 74.20, 33.56, 33.46,
32.91, 32.11, 32.00, 29.80, 29.67, 26.83, 26.17, 25.72, 25.51, 22.98, 22.89, 14.51,
14.37, 10.06, 9.82. HR-MS (MALDI-ToF): m/z ([M]+ ) = 1184.6855, calcd. for C78H92N2O8: m/z = 1184.6853, error = 0.1 ppm. IR: ṽ = 2954, 2924, 2858, 1703, 1194, 744 cm−1
- 13 Osaka I, Zhang R, Sauvé G, Smilgies D.-M, Kowalewski T, McCullough RD. J. Am. Chem. Soc. 2009; 131: 2521