

Subscribe to RSS
DOI: 10.1055/a-2650-6329
Synthesis and Synthetic Applications of Stannylated Amino Acids and Peptides
Authors

Abstract
Amino acids and peptides with stannylated vinyl or aryl side chains are interesting synthetic building blocks that can be easily modified by cross-couplings and metal–halogen exchange reactions, whether for the synthesis of complex natural products and active ingredients or for radiolabeling. The article provides an overview of the new methods and applications.
1
Introduction
Amino acids and peptides play a key role in biological processes in many ways and are involved in many biochemical signaling pathways. Peptide natural products, which are often produced by microorganisms and plants as secondary metabolites, are therefore often important lead structures for the development of active ingredients and drugs.[1] These small peptides differ significantly from larger proteins in that they often contain complex noncanonical amino acids.[2] However, highly active and therefore interesting natural products in particular are often only formed and/or isolated in tiny quantities, and therefore total synthesis is usually the only way to produce sufficient quantities for biological studies. In addition, total synthesis also allows the modification of natural products for studies of the structure–activity relationship (SAR) or for their transformation into tool compounds for chemical biology.[3] Methods are therefore particularly designed for modifying amino acids and peptides and are carried out under the mildest possible conditions, under which, for example, no epimerization of the stereogenic centers of a peptide occurs.[4] In this respect, the Stille coupling[5] is clearly superior to the Suzuki coupling,[6] although organotin compounds are more toxic than the corresponding boronic acids. Nevertheless, aryl and vinyl stannanes are extremely useful synthetic intermediates that can be coupled with a variety of electrophiles often under neutral reaction conditions.[5] In addition, stannylated aromatic amino acids are often used for the synthesis of fluorinated and radiolabeled amino acids and peptides, which find applications as drugs[7] or in medicine, e.g., in positron emission tomography (PET).[8]
This article summarizes the synthesis and applications of stannylated amino acids and peptides and gives an overview of current methods and procedures.
2
Syntheses and Reactions of Amino Acids with Stannylated Aromatic Side Chains
Stannylated aromatic amino acids are often used in the synthesis of fluorinated amino acid derivatives. The incorporation of fluorine often improves the properties of functional molecules. For example, fluorine substituents can increase metabolic stability as well as the speed and extent of penetration of the blood–brain barrier by drugs.[7] Fluorinated aromatic compounds are also used as agrochemicals, materials, and tracers for PET.[8] The latter method, in particular, is an important diagnostic imaging method used to capture essential clinical information.[9] 6-[18F]Fluoro-L-dopa (6-FDOPA) is used, for example, as a probe for central dopamine metabolism in vivo in humans.[10]
The radiofluorodemetallation method is one of the most efficient protocols for the synthesis of radiofluorinated aromatic amino acid probes.[11] Namavari et al. described the regioselective radiofluorodestannylation of 6-trimethylstannyl-L-m-tyrosine and dopa derivatives with F2, [18F]F2, and [18F]acetylhypofluorite.[12] The synthesis of the required stannylated amino acids started from the corresponding aryl iodides such as 1 and their Pd-catalyzed reaction with hexamethyldistannane ([Scheme 1]).[13] With this method, the desired aryltin compounds 2 can be produced regioselectively, while other methods, such as hydrostannylations of in situ generated arines, lead to regioisomeric mixtures.[14] For further conversion, the corresponding fluorinating agent (1%) was passed in a helium stream through a solution of arylstannane 2 in CFCl3 (Freon-11). When [18F]F2 and [18F]acetylhypofluorite were used, the radiochemical yield (RCY) was 23% and 17%, respectively. Acid hydrolysis provided the free fluorinated amino acids. Similar results have also been reported by other groups.[15]

In 2010, the Ritter group described a very efficient method for the late-stage fluorination of complex molecules such as carbohydrates, polyketides, alkaloids, and amino acids.[16] Based on previous work on the electrophilic fluorination of aryl silver compounds,[17] they developed a silver-catalyzed fluorination of aryl stannanes using Selectfluor as a fluorinating agent, which is easier to handle than elemental F2. However, initial trials with AgOTf resulted primarily in protodestannylation products, probably due to the intermediate formation of triflic acid. However, this problem could be solved to a large extent using basic Ag2O. The addition of methanol (5 equiv) also had a positive effect on the yield. In all cases, the degree of protodestannylation could be reduced to less than 10%. The method is suitable for both electron-rich and electron-poor aromatic rings and not only for amino acids, but also for larger peptides such as the enkephalin derivative 4 ([Scheme 2]).

A similar approach was taken by Gouverneur’s group for the synthesis of 18F-labeled dopa by using 18F-selectfluor ditriflat.[18] But again, protodestannylation was the main observed side reaction. However, 18F-labeled dopa could be obtained by this protocol in RCY of about 12% ([Scheme 3]).

Sanford’s group was able to achieve better RCY under Cu-catalyzed conditions.[19] The decisive factor here was the switch to an amide solvent such as DMF or dimethylacetamide and the addition of 15 equiv of pyridine. With [18F]KF, an RCY of around 56% could be achieved in the radiofluorination of a Boc-protected dopa derivative ([Scheme 4]). This method has been applied to the synthesis of a variety of clinically relevant radiotracers and could also be automated.

Further radiofluorinations were described, in which the stannylated amino acids were first converted into the corresponding diaryliodonium salts with Koser reagent,[20] which were then converted with 18F reagents.[21] However, these methods did not result in any improvement in the RCY.
In addition to the production of fluorinated amino acids, stannylated amino acids are also suitable for the generation of complex peptide structures by Stille couplings. An impressive example of this is the synthesis of the biaryl bridge of the right half (blue) of the complestatin molecule ([Fig. 1]).[22] Complestatin was derived from Streptomyces sp. WK-3419 and WK-3490 and is an inhibitor of the gp120-CD4 receptor.[23]

The key steps in the synthesis include the C–C linkage between the central phenylglycine building block and the tryptophan ([Scheme 5]). For this purpose, the diiodinated amino acid 5 was selectively monostannylated, whereby the best results were obtained using (o-tolyl3P)2PdCl2 in the presence of iPr2NEt, although even under these conditions, the yield of 6 was only moderate. iPr2NEt was used to stabilize the resulting Bu3Sn group and to accelerate transmetallation. The iPr2NEt increases the electron-donating property of palladium without disturbing the catalysis cycle.[24] The nevertheless moderate yield is due to the formation of by-products and the acid-lability of the Bu3Sn group. Stille coupling between 6 and the 6-iodotryptophan derivative 7 yielded the desired biaryl compound 8 in 41% yield.

3
Syntheses and Reactions of Amino Acids with Vinylstannane Side Chains
3.1Syntheses of Amino Acids with Vinylstannane Side Chains
3.1.1Syntheses of Amino Acids via Hydrostannylations
In 1992, Crisp and Clink reported on the synthesis of protected stannylated allylglycines via hydrostannylation of propargylglycine 9 ([Scheme 6]).[25] Both radical and catalytic processes were investigated,[26] whereby three differently stannylated amino acid derivatives (E/Z)-10 and 11 became accessible. Under radical conditions, as expected, the terminal products 10 were preferentially obtained, whereby the product distribution depended decisively on the reaction conditions ([Table 1]).[25] Under relatively mild conditions (entry 1), a mixture of (E)-10 and (Z)-10 was obtained with a slight preference for the thermodynamically more stable (E)-product. In thermally induced hydrostannylation (entry 2), on the other hand, only the more stable (E)-product was formed in moderate yield, whether by isomerization or decomposition of (Z)-10. Interestingly, photochemical reaction conditions yielded only traces of 10. As expected, the palladium-catalyzed version (entry 3) provided the products (E)-10 and 11 in a stereospecific syn-addition, which could be separated by chromatography. Unfortunately, this was not possible with (E)-10 and (Z)-10.

In addition to these reactions, the bistannylation of the propargylglycine derivative 9 was also investigated ([Scheme 7]).[25] Reaction of 1 with hexamethylditin in the presence of Pd(PPh3)4 [27] yielded the cis-bis(trimethylstannyl)allylglycine derivative 12 in acceptable yield.

Although the catalytic hydrostannylation processes provide the corresponding syn-addition products in a mechanism-controlled stereospecific manner, the regioselectivity usually leaves much to be desired. Crisp and Gebauer investigated the hydrostannylation of the progargyl glycine derivative 13 with a total of 19 different transition metal complexes, but without achieving a regioselective product formation ([Scheme 8]).[28]

This hydrostannylation process is not limited to terminal alkynes but can also be used with internal alkynes. For example, Isaac et al. reported stereoselective hydrostannylation of aryl-substituted propargylglycines such as 14 but did not address the regioselectivity of the reaction ([Scheme 9]).[29] The products 15 were usually obtained only in moderate yields and were converted directly into the corresponding vinyl iodides.

In connection with the synthesis of photoswitchable glutamate analogues, Isacoff and Trauner et al. used the hydrostannylation of the propargylated pyroglutamic acid derivative 16 for the synthesis of the terminal vinyl stannane 17 ([Scheme 10]), which was subsequently converted into a corresponding azobenzene derivative by Stille coupling.[30] Here, too, the yield in the hydrostannylation step was only moderate; unfortunately, no further information on the regioselectivity was given.

In 2006, Chena and Williams investigated the hydrostannylation of polarized triple bonds as in 18 ([Scheme 11]).[31] Hydride preferentially adds to the β position of the unsaturated ester, causing the tin residue to stand in the α position (19). Attempts to increase the ratio in favor of the β-stannylated product 20 by using sterically more demanding esters were unsuccessful. The corresponding t-butyl ester could no longer be hydrostannylated under the same conditions. However, the corresponding β-stannylated product could selectively be obtained by stannylcupration in the presence of alcohol (see [Scheme 22]).

In contrast to the transition metal–catalyzed hydrostannylations, the corresponding radical reactions provide three possible regioisomers, whereby in terminal alkynes the attack usually takes place preferably at the sterically less hindered position, whereby the more stable vinyl radical is formed. In the case of polyunsaturated compounds, the intermediately formed vinyl radical can be intercepted in cyclization reactions.
For example, Alcaide et al. reported on the synthesis of vinylstannyl-carbapenem 22 by regio- and stereo-controlled intramolecular cyclization of enyne β-lactame 21 ([Scheme 12]).[32] The cyclization process was completely regioselective in all cases. The products were formed by a 5-exo-trig radical process, which is known to be preferred when the radical acceptor has a radical-stabilizing substituent at the β position.[33]

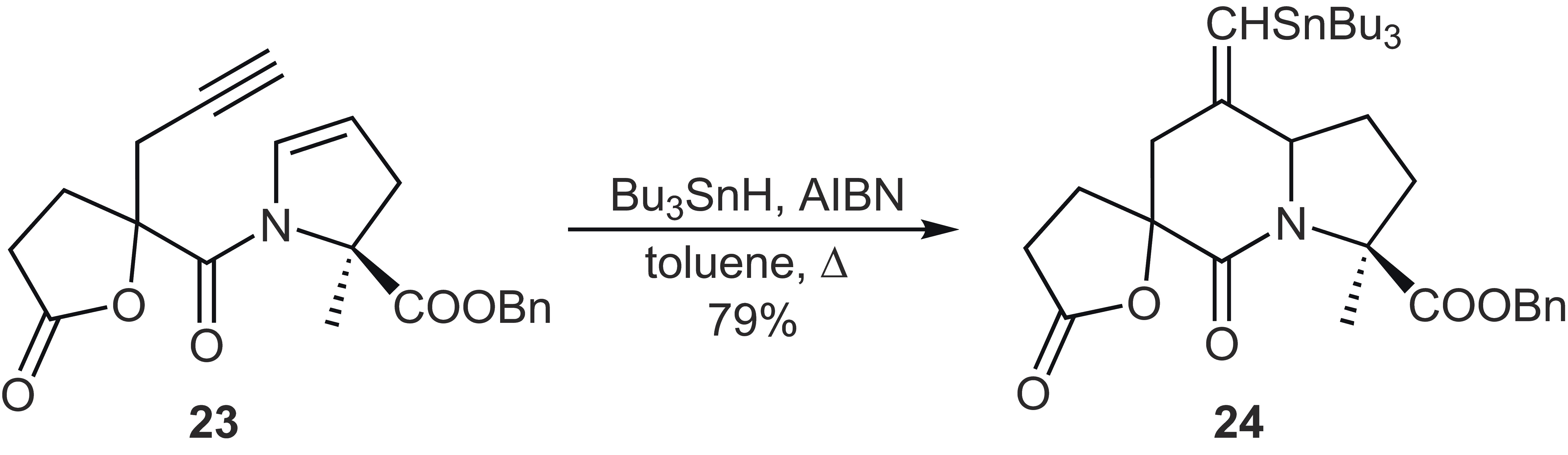
In the context of syntheses of inhibitors of angiotensin converting enzymes, Clivea et al. investigated the radical cyclization of propargylated enamide 23, which was used as a diastereomer mixture of unknown ratios ([Scheme 13]).[34] The cyclization took place without any problems and again yielded only two diastereomers of 24 in a ratio of 3:1, the configuration of which has also not been elucidated. The cyclization was obviously highly stereoselective, as otherwise the formation of further stereoisomers would have been observed. The configuration of the vinyl stannane was also not determined, but rather this group was converted into an exocyclic methylene group by protodestannylation (>90% yield).
3.1.2
Syntheses of Amino Acids by Introduction of Stannylated Side Chains
An alternative approach to the synthesis of γ,δ-unsaturated amino acids uses the highly stereoselective Claisen rearrangement.[35] For example, the deprotonation of N-protected amino acid allyl esters 25 with LDA at −78 °C with the addition of a metal salt such as ZnCl2 presumably leads to the formation of a chelated metal enolate 26, which undergoes a Claisen rearrangement when heated to room temperature, forming γ.δ-unsaturated amino acids 27 ([Scheme 14]).[36] Due to the fixed enolate geometry as a result of chelation and a strong preference for the chair-like transition state, the rearrangement is highly diastereoselective. The use of esters of chiral allylic alcohols allows the highly stereoselective formation of optically active amino acids.[37] Another possibility for asymmetric reaction control is the rearrangement in the presence of chiral ligands.[38] During the rearrangement of peptide allyl esters, the chiral information of the peptide chain can also be used to control the stereochemical outcome of the rearrangement.[39]

To be able to use these protocols for the synthesis of amino acids with a vinylstannane side chain, a regioselective hydrostannylation of allyl alcohol derivatives is required, which preferentially yields the α-stannylated products (R1 = SnR3). The corresponding β-stannylated allyl alcohols (R2 = SnR3) provide allyl stannanes during rearrangement that can no longer be used in Stille couplings. Thus, radical hydrostannylations are ruled out, as they preferentially provide β-products, as well as (E/Z) mixtures.
Chong and coworkers investigated in detail the influence of ligands on the regioselectivity of Pd-catalyzed hydrostannylation of propargyl alcohol derivatives 28 ([Table 2]).[40] When using sterically demanding phosphine ligands such as Cy3P or tBu3P, the β-stannylated product 30 could be obtained with regioselectivities of up to 99:1, but a preferred formation of the α-stannylated allyl alcohol derivatives 29 was not possible. In addition to palladium, however, a number of other transition metals can be used as catalysts.[41] Among other things, Guibe et al. described the application of a molybdenum catalyst.[42] For example, MoBr(allyl)(CO)2(CH3CN)2 is suitable for the hydrostannylation of propargylic alcohol derivatives, but without significant regioselectivity ([Table 2], entries 1–4).
n.r.: not reported.
The catalytically active species in these processes are probably Pd(0) and Mo(0) complexes formed in situ by reduction with tin hydride. Kazmaier et al. therefore also investigated the use of Mo(CO)6 as a catalyst for hydrostannylations. Mo(CO)6 proved to be suitable for this reaction in principle, although the reaction was slow and provided comparatively moderate yields ([Table 2], entry 5).[43] The observed regioselectivities were comparable to the results of Guibe’s catalyst (entries 3 and 4). However, replacing some CO ligands with suitable isonitriles[44] led to a dramatic increase in both yield and selectivity.[45] The best result was achieved with Mo(CO)3(CNt-Bu)3 (MoBI3)[46] (entries 6 and 7); an additional isonitrile ligand had no further positive influence on the reaction.[47]
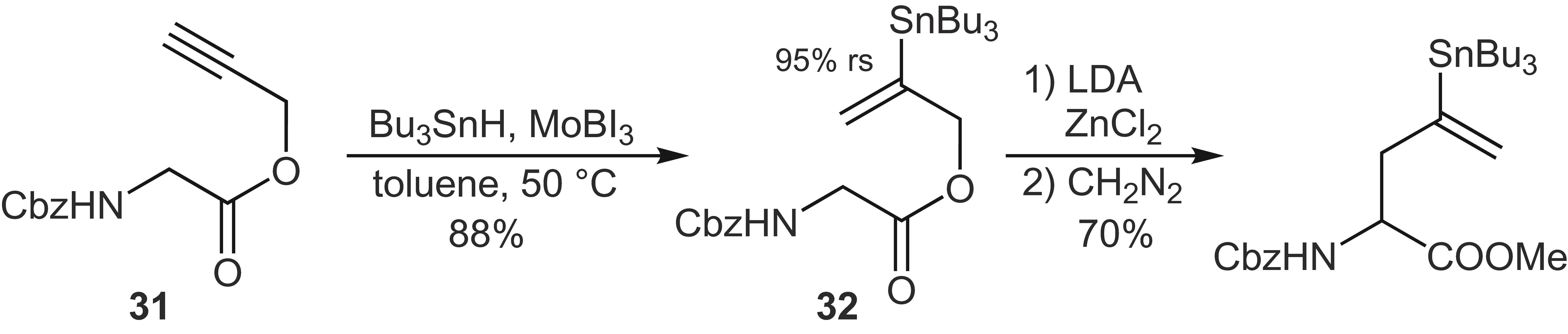
In general, high α-selectivities are achieved with terminal alkynes with this catalyst, even with secondary propargylic alcohols with sterically demanding protective groups. Interestingly, hydrostannylation works not only with allyl alcohols or allyl ethers but also with allyl carboxylates and carbonates. Although MoBI3 has previously been used as a catalyst for allyl alkylations with allyl acetates and carbonates,[48] hydrostannylation seems to take place under such mild conditions that decomposition of the stannylated allyl substrate does not matter.[43] [49] [50] Of particular interest for the synthesis of stannylated amino acids is the direct regioselective hydrostannylation of glycine propargyl esters 31 to the corresponding stannylated allyl esters 32, which can then be rearranged to the stannylated γ,δ-unsaturated amino acids in a chelate enolate-Claisen rearrangement ([Scheme 15]).[51] [52]
The stannylated allyl acetates and carbonates, which are also accessible by hydrostannylation, form another interesting approach to stannylated unsaturated amino acids via palladium-catalyzed allylic alkylation. The chelate enolates already presented during the enolate-Claisen rearrangement are excellently suited as nucleophiles in palladium-catalyzed allylations.[53] These reactions usually only succeed with stabilized enolates, such as malonates, as unstabilized enolates normally coordinate toward the palladium and thus deactivate the catalyst. The few exceptions include tin and zinc ketone enolates and these chelate enolates.[54] Here, the enolate forms a stable chelate complex that does not transmetallate to palladium. At the same time, chelate enolates are highly reactive enolates, which react with π-allyl complexes already at −78 °C. This makes it possible to eliminate even typical side reactions such as the π-σ-π isomerization of the allyl–palladium complexes.[53c] [55] With stannylated allyl acetates and carbonates 33, stannylated amino acid derivatives 34 ([Scheme 16]) can also be obtained under these very mild conditions.[52]

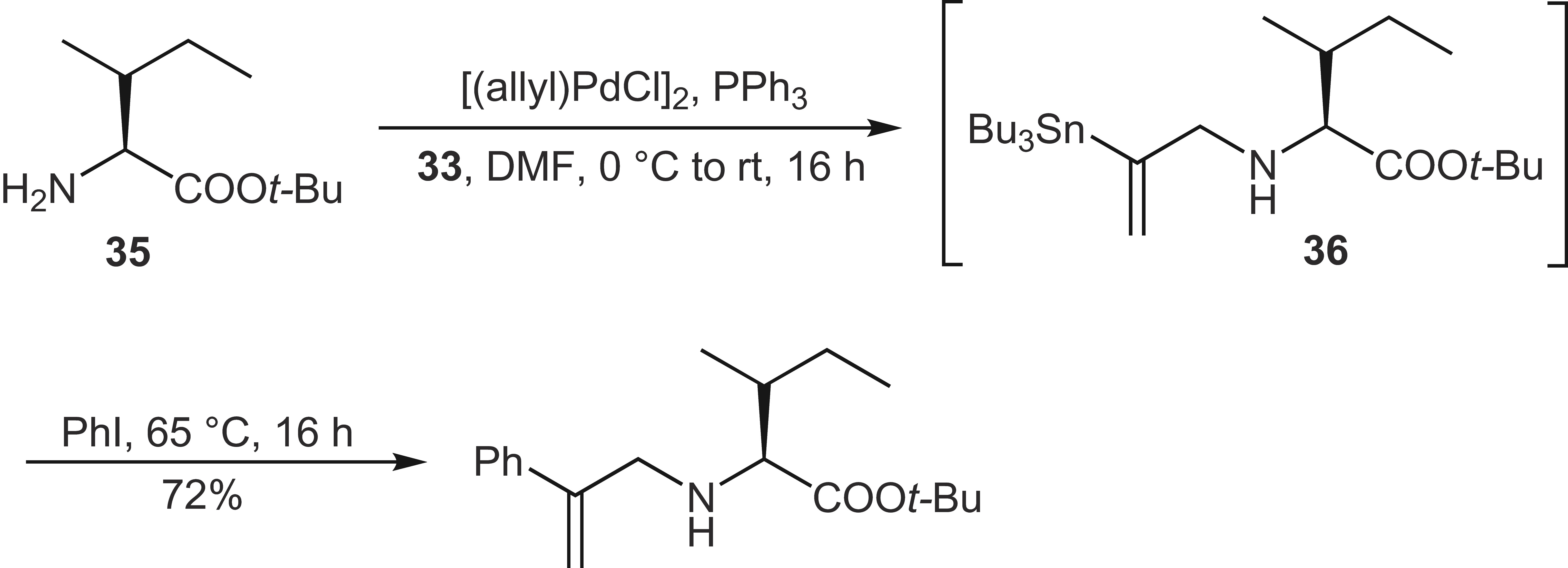
Of course, the “classic” allylation nucleophiles such as amines can also be reacted with these allyl substrates.[56] If, for example, amino acid ester 35 is used as nucleophiles, stannylated N-allylated amino acid derivatives 36 are obtained, which can be further converted directly via Stille couplings in a one-pot process ([Scheme 17]).[57]
In addition to amino acid esters, larger peptide fragments as nucleophiles are also suitable for allylic alkyations.[58] Their deprotonation in the presence of zinc chloride also leads to highly reactive nucleophiles that can be palladium-catalyzed allylated. Excellent diastereoselectivities are achieved almost independently of the allylic substrate used. The stereochemical outcome of the reaction is controlled exclusively by the peptide chain. It is likely that the deprotonated peptide chain coordinates several times to the chelated metal ion. In such metal peptide complexes, one side of the generated enolate is shielded by the side chain of the adjacent amino acid, directing the electrophilic attack to the opposite face of the enolate. This explains that an (S)-amino acid always generates an (R)-amino acid (and vice versa). The method is not limited to C-terminal peptide esters but also works with peptides that contain proline or N-methylated amino acids in the peptide chain. During deprotonation, amide enolates are formed, which are also suitable for palladium-catalyzed allylation.[59] Also functionalized side chains can be installed in a highly stereoselective manner.[60] With stannylated allyl acetates and carbonates such as 33, stannylated peptides 37 are obtained, which allow a variety of further modifications onto the existing peptide framework ([Scheme 18]).[61]

In addition to classic peptides, N-(α-hydroxyacyl)-glycine esters 38 can also be used as nucleophiles.[62] Optimal results are obtained by using ClTi(OiPr)3 as a chelating agent, whereby both diastereomers 39a and 39b can be specifically built up by varying the reaction conditions ([Scheme 19]). Thus, when LDA is used as a base, the (S)/(R) product 39a is preferentially obtained, as with peptides, while LHMDS yields the diastereomeric (S)/(S) product 39b in a highly stereoselective fashion.

3.1.3
Miscellaneous Further Methods
An interesting approach to α-alkylated amino acids with stannylated vinyl side chains was described by Berkowitz et al. ([Scheme 20]).[63] Starting from N-benzoyl-protected (S)-vinylglycine 40, the diastereomeric oxazolines 41a and 41b were obtained by 5-exo-trig-selenocyclization, which could be easily separated chromatographically.[64] Deprotonation of the pure enantiomers and anti-α-alkylation yielded more or less enantiomerically pure oxazolines 42.

The original β,γ-unsaturation was regenerated by base-mediated oxazoline ring opening, whereby this reaction also yielded the corresponding (E)-configured product 43 stereoselectively. By heating with HSnBu3 and AIBN, the corresponding vinyl stannane 44 was obtained, which could be used in different cross-coupling reactions. Protodestannylation yields, for example, the unsubstituted α-alkylated amino acid derivatives 45.
From these, fluorinated stannylated quaternary vinyl amino acids could also be obtained by subjecting 45 to ozonolysis ([Scheme 21]).[65] The formylamino acids 46 formed could be reacted with McCarthy's phosphonate 47,[66] [67] whereby almost exclusively the (E)-fluorovinyl sulfone 48 was formed. This high degree of diastereoselectivity is unusual for condensation reactions of 47 and is probably due to the high steric demand of the quaternary α center. The subsequent sulfone–tin exchange to 49 went smoothly under the mediation of Bu3SnH, usually with very high yields.

Chena and Williams described syntheses of highly functionalized γ,δ-unsaturated amino acids based on a regioselective stannylation of propargylglycine derivatives.[29] Starting from iodoalanine 50, which can be easily obtained from (S)-serine,[68] the corresponding zinc cuprate 51 was generated via halogen–metal exchange, which was then coupled with 3-bromopropiolate ([Scheme 22]).[69] Among the various stannylation methods tested, the addition of [(Me3Sn)CuCN]Li proved to be the method of choice. When 54 was added to [(Me3Sn)CuCN]Li in dry THF at −78 °C, the expected vinyl stannane was obtained as a 1:1 (E/Z) mixture, which unfortunately could not be separated chromatographically. However, if [(Me3Sn)CuCN]Li was treated with EtOH before the addition of 52, only the desired (E)-isomer 20 was formed. The regioisomeric vinylstannane 19 was obtained by hydrostannylation of 52 ([Scheme 11]).

As part of their domoic acid syntheses, the Clayden group investigated the stereoselective formation of trisubstituted double bonds starting from alkinylpyrrolidine 53.[70] Syn-Stannylcupration with [(Bu3Sn)Cu(Bu)CN]Li2 according to the Lipshutz protocol[71] intermediately yielded a vinyl cuprate that was directly methylated ([Scheme 23]). The desired product 54 was the only regio- and stereoisomer obtained. Subsequent Stille couplings provided the desired domoic acids.

As part of their total synthesis of runanins, Kim et al. investigated cross-couplings of the exocyclic bromoalkene group of 55.[72] After all coupling reactions had failed, they converted 55 into vinylstannane 56, which could then be reacted, e.g., with acyl chlorides ([Scheme 24]).

3.2
Reactions of Amino Acids with Vinylstannane Side Chains
Crisp and Clink were the first to investigate the conversion of stannylated amino acids more intensively ([Scheme 25]).[25] For example, copper(II) nitrate–mediated homocoupling reactions[73] of racemic vinylstannanes 11 and (E)-10 gave access to the symmetrically substituted dienes 57 and (E,E)-58 as diastereomer mixtures. Treatment of 11 with a slight excess (1.1 equiv) of iodine yielded the corresponding γ-iodoallylglycine derivative 59, albeit in moderate yield.[74] Interestingly, the corresponding terminal stannane (E)-10 provided not only the expected vinyl iodide (E)-60 but also small amounts (about 6%) of the corresponding (Z)-isomer.

The authors also investigated the modification of stannylated amino acids via Stille couplings.[75] The reaction of 11 with iodobenzene served as a model reaction ([Scheme 26]). Interestingly, the reaction with Pd(PPh3)4 and other palladium catalysts resulted not only in the desired product 61 but also in the unexpected δ-isomers (E)-62 and (Z)-62 resulting from cine substitution. When using more electron-rich ligands such as AsPh3 or P(2-furyl)3, the coupling ran under milder conditions but also delivered more cine product. The cine substitution of vinyl stannanes is usually found only in sterically hindered organostannanes, in which transmetallation occurs only slowly.[76] It is probably the result of a Heck reaction between the terminal alkene position of 11 and the organic electrophile, followed by a palladium-catalyzed elimination of Bu3SnI. For example, no cine substitution product was obtained with the linear vinyl stannane (E)-10, even when the activating ligand AsPh3 was used.

Interestingly, the problem of cine substitution seems to be limited to aryl and hetaryl iodides as well as vinyl halides; with a variety of other electrophiles such as vinyl triflates and allyl and acyl halides, this side reaction has not been observed, even during the use of the activating ligand AsPh3.[51] [52] [75] No isomerization of double bonds was observed in cases where isolated double bonds were formed ([Scheme 27]).

Kazmaier et al. also used the coupling with α,β-unsaturated acid chlorides to build amino acids with heterocyclic side chains by double 1,4-addition of amines ([Scheme 28]).[77]

Stille couplings between vinyl stannanes and vinyl halides or triflates represent an interesting approach to 1,3-dienes, as these couplings occur under neutral conditions and often even at room temperature.[78] These mild conditions allow for the production of delicate dienes that are difficult to obtain by alternative means.[79] Therefore, the formation of the cine product when using vinyl halides is a significant limitation.[75]
Crisp and Gebauer therefore investigated in detail the influence of the N-protection group on the product distribution of the Stille coupling with vinyl bromide.[80] By palladium-catalyzed hydrostannylation of imine-protected propargylglycine ester 13 ([Scheme 8]), the vinyl stannanes 63 and 64 were obtained in a 1:1 ratio, and as such reacted with vinyl bromide in the presence of various catalysts ([Scheme 29]). The relative reactivity of the two isomers was to be investigated. When PdCI2(CH3CN)2 was used as a catalyst with an excess of vinyl bromide, the two coupling products 65 and 66 were formed in a ratio of 5:1, whereby unreacted 64 could be partially recovered. Apparently, the sterically more hindered vinyl stannane reacted more quickly than the terminal one, which may be due to a coordination of the Pd catalyst toward the imine and a chelated-controlled transmetallation. Interestingly, Pd(AsPh3)4 provided the opposite product distribution in a similar ratio. In this case, 63 could be recovered and the reaction was significantly slower. Presumably, the AsPh3 ligand reduces or prevents the coordination of palladium toward the imine nitrogen. The same applies to the acetylated glycine derivative 11, which also preferentially supplied the linear cine substitution product.

For the modification of peptides, Deska and Kazmaier investigated reactions of stannylated peptides such as 37 ([Scheme 30]), which are very easily accessible by palladium-catalyzed allylic alkylation ([Scheme 18]).[61] Very good yields were obtained throughout, with the reaction with some acid chlorides and iodine being approximately quantitative. The vinyl iodide 67 formed in this process could then be further converted in a variety of cross-coupling reactions. Thus, the reaction with vinylstannane also resulted in conjugated diene 68, whereby the problem of cine substitution could be circumvented by this protocol. Carbonylation reactions are suitable for the synthesis of α.β-unsaturated esters and amides (69).[81] Analogous conversions were also achieved with N-(α-hydroxyacyl)-glycine esters.[79]

Junk and Kazmaier also developed a new synthetic route for modified tryptophans based on stannylated amino acids and peptides ([Scheme 31]).[82] Stille couplings of 37 with arbitrarily substituted o-iodanilines yielded styrene derivatives such as 70. Conversion of the free amino functionality into an azide and subsequent irradiation resulted in a C–H insertion of the intermediately formed nitrene and the formation of tryptophans 71. This method was used in the synthesis of several cyclopeptide natural products such as Keramamide A[83] and Mozamide A.[84]

Berkowitz et al. intensively investigated the conversion of α-quaternary stannylated vinylglycine 44 as building blocks for potential applications in de novo peptide design and engineering.[63] The corresponding vinyl stannanes can be produced by radical substitution from the corresponding vinyl selenium compounds ([Scheme 20]), whereby only (E)-configured vinyl stannanes are formed. These can be easily reacted with a large number of electrophiles ([Scheme 32]).[85] Protodestannylation yields the corresponding vinyl amino acids, while when reacted with D2O, the corresponding (E)-configured deuterated amino acids 72 are formed. The reaction with iodine yields the corresponding vinyl iodide 73, which in turn can be converted back into dienes with vinyl stannanes. While 123I-labeled NaI is used to obtain the corresponding radioiodinated amino acids,[86] [87] the analogous radiobrominated derivatives result from the use of NH4 [79]Br.[88]

Starting from γ-fluorinated vinyl stannanes 74, a whole range of fluorinated unsaturated amino acids was obtained in an analogous way, whereby the fluorinated vinyl iodide 75 can in turn be further converted in a series of cross-couplings ([Scheme 33]).[65]

Chena and Williams reported the synthesis of a series of highly functionalized γ,δ-unsaturated amino acids by Stille couplings of stannylated amino acids 19 and 20 ([Scheme 34]).[31] The catalytic system Pd2dba3/CuI/AsPh3 performed quite well for the studied Stille coupling reactions.

The Akiyama group developed an extremely elegant asymmetric one-pot synthesis of dehydroproline esters, based on a formal [3+2] cycloaddition of α-imino esters 76 and allenylstannane 77 ([Scheme 35]).[89] The reaction probably proceeds stepwise via a copper(I)-catalyzed nucleophilic addition of allenylstannane 77 to the α-imino ester 76, forming a vinylic carbenium ion, which ultimately leads to a migration of the tin residue and cyclization. The intermediately formed 4-stannylated dehydroprolinester 78 yielded optically active 4-iodine and 4-aryldehydroproline esters in good yields and high ee’s via iodine oxidation and Stille coupling reactions. Depending on the reaction partner and coupling conditions, the dehydroproline derivative 79, formed by protodestannylation, was found as a by-product, also with good ee’s.

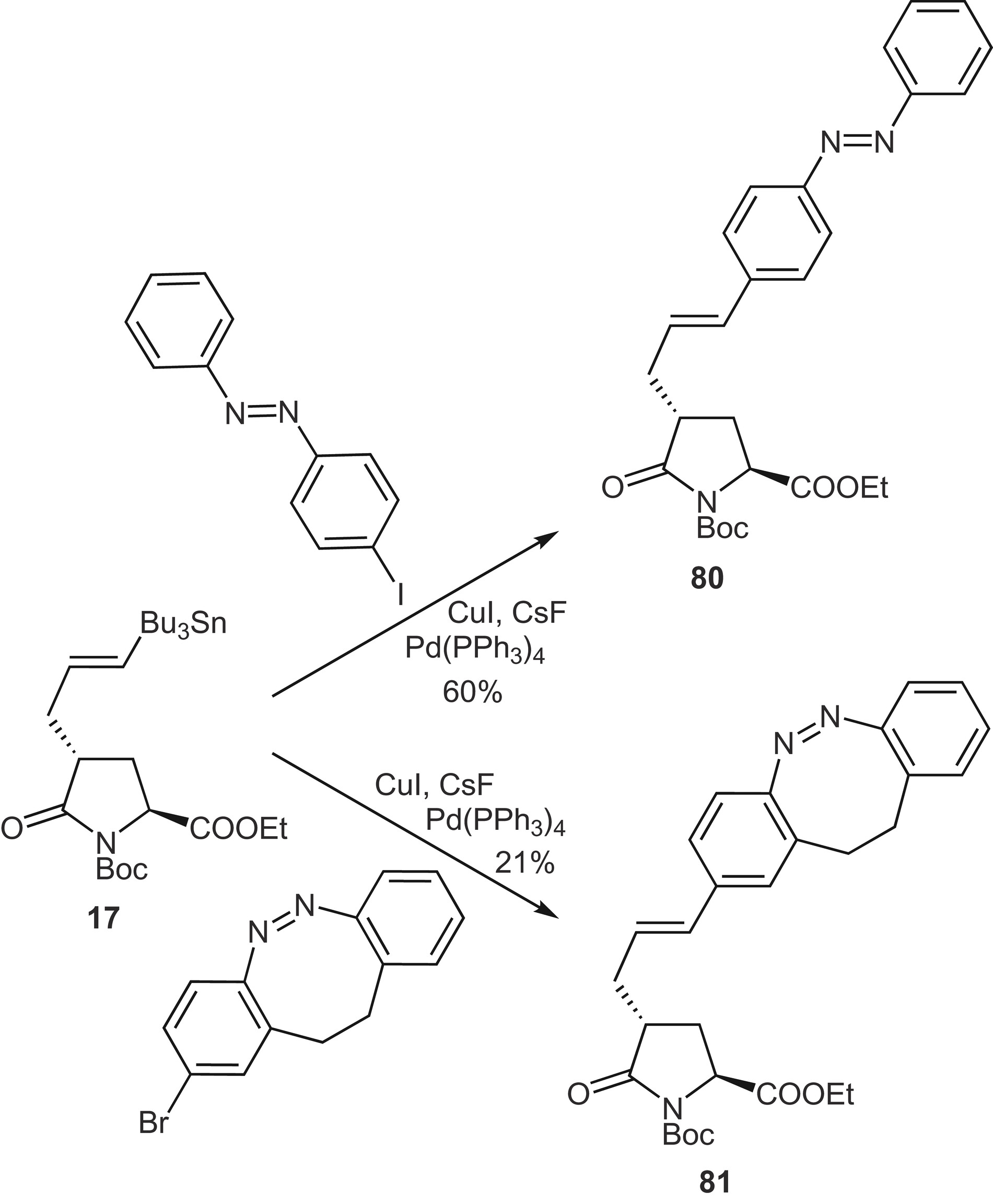
The stannylated pyroglutamic acid derivative 17 was used in the synthesis of photoswitchable glutamate analogues, which are potent and selective iGluR5 and 6-kainate receptor (KAR) agonists ([Scheme 36]). For example, palladium-catalyzed Stille coupling with iodoazobenzene yielded the N-Boc-protected azobenzene pyroglutamate 80.[30] Azobenzenes are the most studied photoswitches and have become popular optical probes for biological systems. The trans-configuration is the thermodynamically preferred form. The cis-configuration is usually metastable, but it can be fixed when an o-bridged azobenzene such as 81 is used.[90]
The Clayden group used vinylstannane 54 as a central building block during the synthesis of kainic acid 82 and various isodomoic acids ([Scheme 37]).[70] Stannane 54 was obtained via stannylcupration and subsequent palladium-catalyzed methylation ([Scheme 23]). Treatment of 54 with TFA resulted in protodestannylation and global deprotection giving rise to 82. The different isodomoic acids were obtained by coupling 54 with different halogenated unsaturated carboxylic acid derivatives and subsequent deprotection.

In the context of the synthesis of Hasubanan alkaloids, Kim et al. described cross-couplings of vinyl stannane 56 with methoxyacetyl chloride, providing exocyclic enone 83 in an acceptable yield ([Scheme 38]).[72]

4
Conclusion
Amino acids and peptides with stannylated vinyl or aryl side chains are interesting synthesis building blocks that can be easily modified by cross-couplings and metal–halogen exchange reactions. Stannylated aromatic amino acids are mainly used for the synthesis of fluorinated and radiolabeled amino acids and peptides, while amino acids with stannylated unsaturated side chains are mainly used in cross-coupling reactions. This allows the synthesis of unusual amino acids and complex natural products. The major advantages of these organotin compounds are the mild and neutral reaction conditions used in their coupling reactions, avoiding epimerization of configurationally labile substrates. The major disadvantage results from the toxicity of many organotin compounds, which is why the cross-coupling reactions should not be carried out at the end of a synthesis, as also the catalysts used often have toxic properties. Recent developments should therefore aim to use more environmentally friendly and nontoxic metals that can be implemented under similarly mild conditions.
Uli Kazmaier
Uli Kazmaier, born in 1960, studied chemistry at the University of Stuttgart where he obtained his diploma (1985) and his PhD (1989). Afterward, he joined as a postdoc in the research groups of M.T. Reetz (Marburg) and B.M. Trost (Stanford). In 1992, he moved to Heidelberg starting his own scientific work as a habilitand at the Institute of Organic Chemistry. In 2000, he received a Novartis Chemistry Lectureship and an offer of a full professorship at the University Bayreuth. In 2001, he also obtained an offer of a full professorship at the University des Saarlandes, which he accepted. His current research interest extends to new organometallic reagents and reactions especially for amino acid and peptide synthesis. Besides the development of new synthetic protocols, the application of these new reactions toward the synthesis of natural products and other pharmaceutical relevant structures plays a central role.

Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgment
I thank Saarland University and Deutsche Forschungsgemeinschaft (DFG) for continuous support of our work and all the highly engaged coworkers for their outstanding contributions.
-
References
- 1a Seebach D, Gardiner J. Acc Chem Res 2008; 41: 1366
- 1b Li G, Lou M, Qi X. Org Chem Front 2022; 9: 517
- 1c Ahmed S, Alam W, Jeandet P. et al. Mar Drugs 2022; 20: 466
- 2a Walsh CT, O‘Brien RV, Khosla C. Angew Chem 2013; 125: 7238
- 2b Walsh CT, O‘Brien RV, Khosla C. Angew Chem Int Ed 2013; 52: 7098
- 3a Burden JE, Davis P, Porreca F, Spatola AF. Bioorg Med Chem Lett 2002; 12: 213
- 3b Weltrowska G, Lemieux C, Chung NN, Schiller PW. J Pept Res 2005; 65: 36
- 3c Rodriguez-Rios M, Megia-Fernandez A, Norman DJ, Bradley M. Chem Soc Rev 2022; 51: 2081
- 4a Lu G-P, Cai C, Lipshutz BH. Green Chem 2013; 15: 105
- 4b Takale BS, Thakore RR, Casotti G, Li X, Gallou F, Lipshutz BH. Angew Chem 2021; 133: 4204
- 4c Takale BS, Thakore RR, Casotti G, Li X, Gallou F, Lipshutz BH. Angew Chem Int Ed 2021; 60: 4158
- 5a Stille JK. Angew Chem 1986; 98: 504
- 5b Stille JK. Angew Chem Int Ed Engl 1986; 25: 508
- 5c Pereyre M, Quintard JP, Rahm A. In: Tin in Organic Synthesis. London: Butterworth; 1987
- 5d Lee V. Org Biomol Chem 2019; 17: 9095
- 6a Suzuki A. Proc Jpn Acad, Ser B 2004; 80: 359
- 6b Lennox AJJ, Lloyd-Jones GC. Chem Soc Rev 2014; 43: 412
- 6c Caso C, Altmann K-H. Chem Eur J 2025; 31: e202402664
- 7 Müller K, Faeh C, Diederich F. Wissenschaft 1881; 2007: 317
- 8a Yamazaki T, Taguchi T, Ojima I. In: Fluorine in Medicinal Chemistry and Chemical Biology. Ojima I. ed. United Kingdom: Wiley-Blackwell; 2009
- 8b Jeschke P. ChemBioChem 2004; 5: 570
- 8c Babudri F, Farinola GM, Naso F, Ragni R. Chem Commun 2007; 1003
- 9 Vallabhajosula S. Molecular Imaging: Radiopharmaceuticals for PET and SPECT. Springer-Verlag; 2009: 5
- 10 Garnett ES, Fimau G, Nahmias C. Nature 1983; 305: 137
- 11 Barrio JR. Neurochem Res 1991; 16: 1047
- 12a Namavari M, Bishop A, Satyamurthy N, Bida G, Barrio JR. Appl Radiat Isot 1992; 43: 989
- 12b Namavari M, Satyamurthy N, Phelps ME, Barrio JR. Appl Radiat Isot 1993; 44: 527
- 13 Azizian H, Eaborn C, Pidcock AJ. Organometal Chem 1981; 215: 49
- 14 Lakshmi BV, Wefelscheid UK, Kazmaier U. Synlett 2011; 345
- 15a Hess E, Sichler S, Kluge A, Coenen HH. Appl Radiat Isot 2002; 57: 185
- 15b Füchtner F, Steinbach J. Appl Radiat Isot 2003; 58: 575
- 16 Tang P, Furuya T, Ritter T. J Am Chem Soc 2010; 132: 12150
- 17a Furuya T, Strom AE, Ritter T. J Am Chem Soc 2009; 131: 1662
- 17b Furuya T, Ritter T. Org Lett 2009; 11: 2860
- 18 Stenhagen ISR, Kirjavainen AK, Forsback SJ. et al. Chem Commun 2013; 49: 1386
- 19 Makaravage KJ, Brooks AF, Mossine AV, Sanford MS, Scott PJH. Org Lett 2016; 18: 5440
- 20a Kirschning A. Eur J Org Chem 1998; 2267
- 20b Kamal R, Omkar Kumar V, Kumar R. Chemistryselect 2022; 7: e202103719
- 21a Yusubov MS, Svitich Y, Larkina MS, Zhdankin VV. ARKIVOC 2013; 364
- 21b Tredwell M, Gouverneur V. Angew Chem 2012; 124: 11590
- 21c Tredwell M, Gouverneur V. Angew Chem Int Ed 2012; 51: 11426
- 21d Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Nat Commun 2014; 5: 4365
- 21e Maisonial-Besset A, Serre A, Ouadi A. et al. Eur J Org Chem 2018; 7058
- 21f Edwards W, de Vries W, Westwell AD, Daniels S, Wirth T. Eur J Org Chem 2015; 6909
- 22 Yamada Y, Akiba A, Arima S. et al. Chem Pharm Bull 2005; 53: 1277
- 23a Kaneko I, Fearon DT, Austen K. J Immunol 1980; 124: 1194
- 23b Kaneko I, Kamoshida K, Takahashi S. J Antibiot 1989; 42: 236
- 23c Matsuzaki K, Ikeda H, Ogino T. et al. J Antibiot 1994; 47: 1173
- 24 Kai T, Kajimoto N, Konda Yamada Y, Harigaya Y, Takayanagi H. Tetrahedron Lett 1999; 40: 6289
- 25 Crisp GT, Clink PT. Tetrahedron Lett 1992; 33: 4649
- 26a Collins PW, Jung CJ, Gasiecki A, Pappo R. Tetrahedron Lett 1978; 19: 3187
- 26b Jung ME, Light LA. Tetrahedron Lett 1982; 23: 3851
- 26c Nozaki K, Oshima K, Utimoto K. Tetrahedron 1989; 45: 923
- 27a Mitchell TN, Amamria A, Killing H, Rutschow D. J Organomet Chem 1986; 304: 257
- 27b Piers E, Skerlj RT. J Chem Soc, Chem Commun 1986; 626
- 28 Crisp GT, Gebauer MG. J Organomet Chem 1997; 532: 83
- 29 Isaac M, Slassi A, Da Silva K, Xin T. Tetrahedron Lett 2001; 42: 2957
- 30 Volgraf M, Gorostiza P, Szobota S, Helix MR, Isacoff EY, Trauner D. J Am Chem Soc 2007; 129: 260
- 31 Chena S, Williams RM. Tetrahedron 2006; 62: 11572
- 32a Alcaide B, Benito JL, Rodríguez-Campos IM. et al. Tetrahedron Asymmetry 1995; 6: 1055
- 32b Alcaide B, Rodríguez-Campos IM, Rodríguez-López J, Rodríguez-Vicente A. J Org Chem 1999; 64: 5377
- 33 Baldwin JE. J Chem Soc, Chem Commun 1976; 734
- 34 Clive DLJ, Yang HK, Lewanczuk RZ. Compt Rend Acad Sci Chem 2001; 4: 505
- 37a Kazmaier U, Schneider C. Tetrahedron Lett 1998; 39: 817
- 37b Kazmaier U, Schneider C. Synthesis 1998; 1321
- 38a Kazmaier U, Krebs A. Angew Chem 1995; 107: 2213
- 38b Kazmaier U, Krebs A. Angew Chem Int Ed Engl 1995; 34: 2012
- 38c Krebs A, Kazmaier U. Tetrahedron Lett 1996; 37: 7945
- 38d Kazmaier U, Krebs A. Tetrahedron Lett 1999; 40: 479
- 38e Mues H, Kazmaier U. Synthesis 2001; 487
- 38f Kazmaier U, Mues H, Krebs A. Chem Eur J 1850; 2002: 8
- 39a Kazmaier U. J Org Chem 1994; 59: 6667
- 39b Kazmaier U, Maier S. J Chem Soc, Chem Commun 1998; 2535
- 39c Kazmaier U, Maier S. J Org Chem 1999; 64: 4574
- 40 Darwish A, Lang A, Kim T, Chong JM. Org Lett 2008; 10: 861
- 41 Kikukawa K, Umekawa H, Wada F, Matsuda T. Chem Lett 1988; 881
- 42 Zhang HX, Guibe F, Balavoine G. J Org Chem 1867; 1990: 55
- 43 Kazmaier U, Pohlman M, Schauβ D. Eur J Org Chem 2000; 2761
- 44 Hegedus LS. Organic Synthesis with Transition Metals. Weinheim: VCH; 1995
- 45 Kazmaier U, Schauβ D, Pohlman M. Org Lett 1999; 1: 1017
- 46a Albers MO, Coville NJ, Ashworth TV, Singleton E, Swanepoel HE. J Organomet Chem 1980; 199: 55
- 46b Albers MO, Coville NJ. Coord Chem Rev 1984; 53: 227
- 47 Braune S, Kazmaier U. J Organomet Chem 2002; 641: 26
- 48 Trost BM, Merlic CA. J Am Chem Soc 1990; 112: 9590
- 49 Wesquet AO, Dörrenbächer S, Kazmaier U. Synlett 2006; 1105
- 50 Kazmaier U, Dörrenbächer S, Wesquet A, Lucas S, Kummeter M. Synthesis 2007; 320
- 51 Kazmaier U, Schauβ D, Pohlman M, Raddatz S. Synthesis 2000; 914
- 52 Kazmaier U, Schauβ D, Raddatz S, Pohlman M. Chem Eur J 2001; 7: 456
- 53a Kazmaier U, Zumpe FL. Angew Chem 1999; 111: 1572
- 53b Kazmaier U, Zumpe FL. Angew Chem Int Ed Engl 1999; 38: 1468
- 53c Kazmaier U, Maier S, Zumpe FL. Synlett 2000; 1523
- 53d Kazmaier U, Zumpe FL. Eur J Org Chem 2001; 4067
- 53e Lindner T, Kazmaier U. Adv Synth Catal 2005; 347: 1687
- 53f Kazmaier U, Lindner T. Angew Chem 2005; 117: 3368
- 53g Kazmaier U, Lindner T. Angew Chem Int Ed 2005; 44: 3303
- 54a Kazmaier U. Curr Org Chem 2003; 317
- 54b Kazmaier UJ. Ind Chem Soc 2003; 80: 957
- 54c Huwig K, Schultz K, Kazmaier U. Angew Chem 2015; 127: 9248
- 54d Huwig K, Schultz K, Kazmaier U. Angew Chem Int Ed 2015; 54: 9120
- 54e Kazmaier U. Org Chem Front 2016; 3: 1541
- 54f Junk L, Kazmaier U. ChemistryOpen 2020; 9: 929
- 55a Kazmaier U, Zumpe FL. Angew Chem 2000; 112: 805
- 55b Kazmaier U, Zumpe FL. Angew Chem Int Ed Engl 2000; 39: 802
- 55c Kazmaier U, Pohlman M. Synlett 2004; 623
- 55d Krämer K, Kazmaier U. J Org Chem 2006; 71: 8950
- 56a Bukovec C, Wesquet AO, Kazmaier U. Eur J Org Chem 2011; 1047
- 56b Bukovec C, Kazmaier U. Org Biomol Chem 2011; 9: 2743
- 57 Bukovec C, Kazmaier U. Org Lett 2009; 11: 3518
- 58a Kazmaier U, Deska J, Watzke A. Angew Chem 2006; 118: 4973
- 58b Kazmaier U, Deska J, Watzke A. Angew Chem Int Ed 2006; 45: 4855
- 58c Deska J, Kazmaier U. Chem Eur J 2007; 13: 6204
- 58d Kazmaier U, Bayer A, Deska J. Synthesis 2013; 45: 1462
- 59a Datta S, Kazmaier U. Org Biomol Chem 2011; 9: 872
- 59b Datta S, Bayer A, Kazmaier U. Org Biomol Chem 2012; 10: 8268
- 60a Krämer K, Deska J, Hebach C, Kazmaier U. Org Biomol Chem 2009; 7: 103
- 60b Servatius P, Kazmaier U. J Org Chem 2018; 83: 11341
- 61a Deska J, Kazmaier U. Angew Chem 2007; 119: 4570
- 61b Deska J, Kazmaier U. Angew Chem Int Ed 2007; 46: 4570
- 62 Horn A, Kazmaier U. Org Lett 2019; 21: 4595
- 63 Berkowitz DB, McFadden JM, Chisowa E, Semerad CL. J Am Chem Soc 2000; 122: 11031
- 64 Berkowitz DB, Smith MK. Synthesis 1996; 39
- 65a Berkowitz DB, De La Salud-Bea R, Jahng W-J. Org Lett 1821; 2004: 6
- 65b McCune CD, Beio ML, Sturdivant JM, De La Salud-Bea R, Darnell BM, Berkowitz DB. J Am Chem Soc 2017; 139: 14077
- 66a McCarthy JR, Matthews DP, Stemerick DM. et al. J Am Chem Soc 1991; 113: 7439
- 66b McCarthy JR, Matthews DP, Paolini JP. Org Synth 1995; 72: 216
- 67 McCarthy JR, Huber EW, Le T-B, Laskovics FM, Matthews DP. Tetrahedron 1996; 52: 45
- 68 Jackson RFW, Perez-Gonzalez M. Org Synth 2005; 81: 77
- 69a Dunn MJ, Jackson RFW, Pietruszka J, Turner D. J Org Chem 1995; 60: 2210
- 69b Reiβer M, Maas G. Synthesis 1998; 1129
- 70 Lemière G, Sedehizadeh S, Toueg J, Fleary-Roberts N, Clayden J. Chem Commun 2011; 47: 3745
- 71a Lipshutz BH, Ellsworth EL, Dimock SH, Reuter DC. Tetrahedron Lett 1989; 30: 2065
- 71b Gopalarathnam A, Nelson SG. Org Lett 2006; 8: 7
- 71c Liang G, Seiple IB, Trauner D. Org Lett 2005; 7: 2837
- 71d Harris L, Jarowicki P, Kocienski P, Bell R. Synlett 1996; 903
- 72 Tan S, Li F, Park S, Kim S. Org Lett 2019; 21: 292-295
- 73 Ghosal S, Lukas GP, Kyler KS. J Org Chem 1987; 52: 4296
- 74 Crisp GT, Robertson TA. Tetrahedron 1992; 48: 3239
- 75 Crisp GT, Glink PT. Tetrahedron 1994; 50: 3213
- 76a Kikukawa K, Umekaw H, Matsuda T. J Organomet Chem 1986; 311: C44
- 76b Storch G, Richard CA. J Am Chem Soc 1990; 112: 7399
- 76c Peng Y, Li W-DZ. Eur J Org Chem 2010; 35: 6703
- 77 Dörrenbächer S, Kazmaier U, Ruf S. Synlett 2006; 547
- 78a Moriarty RM, Epa WR. Tetrahedron Lett 1992; 33: 4095
- 78b Beletskaya IP. J Organomet Chem 1983; 250: 551
- 79 Nicolaou KC, Chakraborty TK, Piscopio AD, Minowa N, Bertinato P. J Am Chem Soc 1993; 115: 4419
- 80 Crisp GT, Gebauer MG. Tetrahedron Lett 1995; 36: 3389
- 81 Servatius P, Junk L, Kazmaier U. Synlett 2019; 30: 1289
- 83a Junk L, Kazmaier U. Angew Chem 2018; 130: 11602
- 83b Junk L, Kazmaier U. Angew Chem Int Ed 2018; 57: 11432
- 84 Junk L, Kazmaier U. J Org Chem 2019; 84: 2489
- 85 Berkowitz DB, Chisowa E, McFadden JM. Tetrahedron 2001; 57: 6329
- 86 Yu W, McConathy J, Olson J, Lager VM, Goodman MM. J Med Chem 2007; 50: 6718
- 87 Yu W, Williams L, Malveaux E, Camp VM, Olson J, Goodman MM. Bioorg Med Chem Lett 2008; 18: 1264
- 88 Bartels JL, Huang C, Li A. et al. J Med Chem 2015; 58: 8542
- 89 Fuchibe K, Hatemata R, Akiyama T. Tetrahedron 2006; 62: 11304
- 90 Thapaliya ER, Zhao J, Ellis-Davies GCR. ACS Chem Neurosci 2019; 10: 2481
Correspondence
Publication History
Received: 17 May 2025
Accepted after revision: 03 July 2025
Accepted Manuscript online:
07 July 2025
Article published online:
20 August 2025
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/).
Georg Thieme Verlag KG
Oswald-Hesse-Straβe 50, 70469 Stuttgart, Germany
-
References
- 1a Seebach D, Gardiner J. Acc Chem Res 2008; 41: 1366
- 1b Li G, Lou M, Qi X. Org Chem Front 2022; 9: 517
- 1c Ahmed S, Alam W, Jeandet P. et al. Mar Drugs 2022; 20: 466
- 2a Walsh CT, O‘Brien RV, Khosla C. Angew Chem 2013; 125: 7238
- 2b Walsh CT, O‘Brien RV, Khosla C. Angew Chem Int Ed 2013; 52: 7098
- 3a Burden JE, Davis P, Porreca F, Spatola AF. Bioorg Med Chem Lett 2002; 12: 213
- 3b Weltrowska G, Lemieux C, Chung NN, Schiller PW. J Pept Res 2005; 65: 36
- 3c Rodriguez-Rios M, Megia-Fernandez A, Norman DJ, Bradley M. Chem Soc Rev 2022; 51: 2081
- 4a Lu G-P, Cai C, Lipshutz BH. Green Chem 2013; 15: 105
- 4b Takale BS, Thakore RR, Casotti G, Li X, Gallou F, Lipshutz BH. Angew Chem 2021; 133: 4204
- 4c Takale BS, Thakore RR, Casotti G, Li X, Gallou F, Lipshutz BH. Angew Chem Int Ed 2021; 60: 4158
- 5a Stille JK. Angew Chem 1986; 98: 504
- 5b Stille JK. Angew Chem Int Ed Engl 1986; 25: 508
- 5c Pereyre M, Quintard JP, Rahm A. In: Tin in Organic Synthesis. London: Butterworth; 1987
- 5d Lee V. Org Biomol Chem 2019; 17: 9095
- 6a Suzuki A. Proc Jpn Acad, Ser B 2004; 80: 359
- 6b Lennox AJJ, Lloyd-Jones GC. Chem Soc Rev 2014; 43: 412
- 6c Caso C, Altmann K-H. Chem Eur J 2025; 31: e202402664
- 7 Müller K, Faeh C, Diederich F. Wissenschaft 1881; 2007: 317
- 8a Yamazaki T, Taguchi T, Ojima I. In: Fluorine in Medicinal Chemistry and Chemical Biology. Ojima I. ed. United Kingdom: Wiley-Blackwell; 2009
- 8b Jeschke P. ChemBioChem 2004; 5: 570
- 8c Babudri F, Farinola GM, Naso F, Ragni R. Chem Commun 2007; 1003
- 9 Vallabhajosula S. Molecular Imaging: Radiopharmaceuticals for PET and SPECT. Springer-Verlag; 2009: 5
- 10 Garnett ES, Fimau G, Nahmias C. Nature 1983; 305: 137
- 11 Barrio JR. Neurochem Res 1991; 16: 1047
- 12a Namavari M, Bishop A, Satyamurthy N, Bida G, Barrio JR. Appl Radiat Isot 1992; 43: 989
- 12b Namavari M, Satyamurthy N, Phelps ME, Barrio JR. Appl Radiat Isot 1993; 44: 527
- 13 Azizian H, Eaborn C, Pidcock AJ. Organometal Chem 1981; 215: 49
- 14 Lakshmi BV, Wefelscheid UK, Kazmaier U. Synlett 2011; 345
- 15a Hess E, Sichler S, Kluge A, Coenen HH. Appl Radiat Isot 2002; 57: 185
- 15b Füchtner F, Steinbach J. Appl Radiat Isot 2003; 58: 575
- 16 Tang P, Furuya T, Ritter T. J Am Chem Soc 2010; 132: 12150
- 17a Furuya T, Strom AE, Ritter T. J Am Chem Soc 2009; 131: 1662
- 17b Furuya T, Ritter T. Org Lett 2009; 11: 2860
- 18 Stenhagen ISR, Kirjavainen AK, Forsback SJ. et al. Chem Commun 2013; 49: 1386
- 19 Makaravage KJ, Brooks AF, Mossine AV, Sanford MS, Scott PJH. Org Lett 2016; 18: 5440
- 20a Kirschning A. Eur J Org Chem 1998; 2267
- 20b Kamal R, Omkar Kumar V, Kumar R. Chemistryselect 2022; 7: e202103719
- 21a Yusubov MS, Svitich Y, Larkina MS, Zhdankin VV. ARKIVOC 2013; 364
- 21b Tredwell M, Gouverneur V. Angew Chem 2012; 124: 11590
- 21c Tredwell M, Gouverneur V. Angew Chem Int Ed 2012; 51: 11426
- 21d Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Nat Commun 2014; 5: 4365
- 21e Maisonial-Besset A, Serre A, Ouadi A. et al. Eur J Org Chem 2018; 7058
- 21f Edwards W, de Vries W, Westwell AD, Daniels S, Wirth T. Eur J Org Chem 2015; 6909
- 22 Yamada Y, Akiba A, Arima S. et al. Chem Pharm Bull 2005; 53: 1277
- 23a Kaneko I, Fearon DT, Austen K. J Immunol 1980; 124: 1194
- 23b Kaneko I, Kamoshida K, Takahashi S. J Antibiot 1989; 42: 236
- 23c Matsuzaki K, Ikeda H, Ogino T. et al. J Antibiot 1994; 47: 1173
- 24 Kai T, Kajimoto N, Konda Yamada Y, Harigaya Y, Takayanagi H. Tetrahedron Lett 1999; 40: 6289
- 25 Crisp GT, Clink PT. Tetrahedron Lett 1992; 33: 4649
- 26a Collins PW, Jung CJ, Gasiecki A, Pappo R. Tetrahedron Lett 1978; 19: 3187
- 26b Jung ME, Light LA. Tetrahedron Lett 1982; 23: 3851
- 26c Nozaki K, Oshima K, Utimoto K. Tetrahedron 1989; 45: 923
- 27a Mitchell TN, Amamria A, Killing H, Rutschow D. J Organomet Chem 1986; 304: 257
- 27b Piers E, Skerlj RT. J Chem Soc, Chem Commun 1986; 626
- 28 Crisp GT, Gebauer MG. J Organomet Chem 1997; 532: 83
- 29 Isaac M, Slassi A, Da Silva K, Xin T. Tetrahedron Lett 2001; 42: 2957
- 30 Volgraf M, Gorostiza P, Szobota S, Helix MR, Isacoff EY, Trauner D. J Am Chem Soc 2007; 129: 260
- 31 Chena S, Williams RM. Tetrahedron 2006; 62: 11572
- 32a Alcaide B, Benito JL, Rodríguez-Campos IM. et al. Tetrahedron Asymmetry 1995; 6: 1055
- 32b Alcaide B, Rodríguez-Campos IM, Rodríguez-López J, Rodríguez-Vicente A. J Org Chem 1999; 64: 5377
- 33 Baldwin JE. J Chem Soc, Chem Commun 1976; 734
- 34 Clive DLJ, Yang HK, Lewanczuk RZ. Compt Rend Acad Sci Chem 2001; 4: 505
- 37a Kazmaier U, Schneider C. Tetrahedron Lett 1998; 39: 817
- 37b Kazmaier U, Schneider C. Synthesis 1998; 1321
- 38a Kazmaier U, Krebs A. Angew Chem 1995; 107: 2213
- 38b Kazmaier U, Krebs A. Angew Chem Int Ed Engl 1995; 34: 2012
- 38c Krebs A, Kazmaier U. Tetrahedron Lett 1996; 37: 7945
- 38d Kazmaier U, Krebs A. Tetrahedron Lett 1999; 40: 479
- 38e Mues H, Kazmaier U. Synthesis 2001; 487
- 38f Kazmaier U, Mues H, Krebs A. Chem Eur J 1850; 2002: 8
- 39a Kazmaier U. J Org Chem 1994; 59: 6667
- 39b Kazmaier U, Maier S. J Chem Soc, Chem Commun 1998; 2535
- 39c Kazmaier U, Maier S. J Org Chem 1999; 64: 4574
- 40 Darwish A, Lang A, Kim T, Chong JM. Org Lett 2008; 10: 861
- 41 Kikukawa K, Umekawa H, Wada F, Matsuda T. Chem Lett 1988; 881
- 42 Zhang HX, Guibe F, Balavoine G. J Org Chem 1867; 1990: 55
- 43 Kazmaier U, Pohlman M, Schauβ D. Eur J Org Chem 2000; 2761
- 44 Hegedus LS. Organic Synthesis with Transition Metals. Weinheim: VCH; 1995
- 45 Kazmaier U, Schauβ D, Pohlman M. Org Lett 1999; 1: 1017
- 46a Albers MO, Coville NJ, Ashworth TV, Singleton E, Swanepoel HE. J Organomet Chem 1980; 199: 55
- 46b Albers MO, Coville NJ. Coord Chem Rev 1984; 53: 227
- 47 Braune S, Kazmaier U. J Organomet Chem 2002; 641: 26
- 48 Trost BM, Merlic CA. J Am Chem Soc 1990; 112: 9590
- 49 Wesquet AO, Dörrenbächer S, Kazmaier U. Synlett 2006; 1105
- 50 Kazmaier U, Dörrenbächer S, Wesquet A, Lucas S, Kummeter M. Synthesis 2007; 320
- 51 Kazmaier U, Schauβ D, Pohlman M, Raddatz S. Synthesis 2000; 914
- 52 Kazmaier U, Schauβ D, Raddatz S, Pohlman M. Chem Eur J 2001; 7: 456
- 53a Kazmaier U, Zumpe FL. Angew Chem 1999; 111: 1572
- 53b Kazmaier U, Zumpe FL. Angew Chem Int Ed Engl 1999; 38: 1468
- 53c Kazmaier U, Maier S, Zumpe FL. Synlett 2000; 1523
- 53d Kazmaier U, Zumpe FL. Eur J Org Chem 2001; 4067
- 53e Lindner T, Kazmaier U. Adv Synth Catal 2005; 347: 1687
- 53f Kazmaier U, Lindner T. Angew Chem 2005; 117: 3368
- 53g Kazmaier U, Lindner T. Angew Chem Int Ed 2005; 44: 3303
- 54a Kazmaier U. Curr Org Chem 2003; 317
- 54b Kazmaier UJ. Ind Chem Soc 2003; 80: 957
- 54c Huwig K, Schultz K, Kazmaier U. Angew Chem 2015; 127: 9248
- 54d Huwig K, Schultz K, Kazmaier U. Angew Chem Int Ed 2015; 54: 9120
- 54e Kazmaier U. Org Chem Front 2016; 3: 1541
- 54f Junk L, Kazmaier U. ChemistryOpen 2020; 9: 929
- 55a Kazmaier U, Zumpe FL. Angew Chem 2000; 112: 805
- 55b Kazmaier U, Zumpe FL. Angew Chem Int Ed Engl 2000; 39: 802
- 55c Kazmaier U, Pohlman M. Synlett 2004; 623
- 55d Krämer K, Kazmaier U. J Org Chem 2006; 71: 8950
- 56a Bukovec C, Wesquet AO, Kazmaier U. Eur J Org Chem 2011; 1047
- 56b Bukovec C, Kazmaier U. Org Biomol Chem 2011; 9: 2743
- 57 Bukovec C, Kazmaier U. Org Lett 2009; 11: 3518
- 58a Kazmaier U, Deska J, Watzke A. Angew Chem 2006; 118: 4973
- 58b Kazmaier U, Deska J, Watzke A. Angew Chem Int Ed 2006; 45: 4855
- 58c Deska J, Kazmaier U. Chem Eur J 2007; 13: 6204
- 58d Kazmaier U, Bayer A, Deska J. Synthesis 2013; 45: 1462
- 59a Datta S, Kazmaier U. Org Biomol Chem 2011; 9: 872
- 59b Datta S, Bayer A, Kazmaier U. Org Biomol Chem 2012; 10: 8268
- 60a Krämer K, Deska J, Hebach C, Kazmaier U. Org Biomol Chem 2009; 7: 103
- 60b Servatius P, Kazmaier U. J Org Chem 2018; 83: 11341
- 61a Deska J, Kazmaier U. Angew Chem 2007; 119: 4570
- 61b Deska J, Kazmaier U. Angew Chem Int Ed 2007; 46: 4570
- 62 Horn A, Kazmaier U. Org Lett 2019; 21: 4595
- 63 Berkowitz DB, McFadden JM, Chisowa E, Semerad CL. J Am Chem Soc 2000; 122: 11031
- 64 Berkowitz DB, Smith MK. Synthesis 1996; 39
- 65a Berkowitz DB, De La Salud-Bea R, Jahng W-J. Org Lett 1821; 2004: 6
- 65b McCune CD, Beio ML, Sturdivant JM, De La Salud-Bea R, Darnell BM, Berkowitz DB. J Am Chem Soc 2017; 139: 14077
- 66a McCarthy JR, Matthews DP, Stemerick DM. et al. J Am Chem Soc 1991; 113: 7439
- 66b McCarthy JR, Matthews DP, Paolini JP. Org Synth 1995; 72: 216
- 67 McCarthy JR, Huber EW, Le T-B, Laskovics FM, Matthews DP. Tetrahedron 1996; 52: 45
- 68 Jackson RFW, Perez-Gonzalez M. Org Synth 2005; 81: 77
- 69a Dunn MJ, Jackson RFW, Pietruszka J, Turner D. J Org Chem 1995; 60: 2210
- 69b Reiβer M, Maas G. Synthesis 1998; 1129
- 70 Lemière G, Sedehizadeh S, Toueg J, Fleary-Roberts N, Clayden J. Chem Commun 2011; 47: 3745
- 71a Lipshutz BH, Ellsworth EL, Dimock SH, Reuter DC. Tetrahedron Lett 1989; 30: 2065
- 71b Gopalarathnam A, Nelson SG. Org Lett 2006; 8: 7
- 71c Liang G, Seiple IB, Trauner D. Org Lett 2005; 7: 2837
- 71d Harris L, Jarowicki P, Kocienski P, Bell R. Synlett 1996; 903
- 72 Tan S, Li F, Park S, Kim S. Org Lett 2019; 21: 292-295
- 73 Ghosal S, Lukas GP, Kyler KS. J Org Chem 1987; 52: 4296
- 74 Crisp GT, Robertson TA. Tetrahedron 1992; 48: 3239
- 75 Crisp GT, Glink PT. Tetrahedron 1994; 50: 3213
- 76a Kikukawa K, Umekaw H, Matsuda T. J Organomet Chem 1986; 311: C44
- 76b Storch G, Richard CA. J Am Chem Soc 1990; 112: 7399
- 76c Peng Y, Li W-DZ. Eur J Org Chem 2010; 35: 6703
- 77 Dörrenbächer S, Kazmaier U, Ruf S. Synlett 2006; 547
- 78a Moriarty RM, Epa WR. Tetrahedron Lett 1992; 33: 4095
- 78b Beletskaya IP. J Organomet Chem 1983; 250: 551
- 79 Nicolaou KC, Chakraborty TK, Piscopio AD, Minowa N, Bertinato P. J Am Chem Soc 1993; 115: 4419
- 80 Crisp GT, Gebauer MG. Tetrahedron Lett 1995; 36: 3389
- 81 Servatius P, Junk L, Kazmaier U. Synlett 2019; 30: 1289
- 83a Junk L, Kazmaier U. Angew Chem 2018; 130: 11602
- 83b Junk L, Kazmaier U. Angew Chem Int Ed 2018; 57: 11432
- 84 Junk L, Kazmaier U. J Org Chem 2019; 84: 2489
- 85 Berkowitz DB, Chisowa E, McFadden JM. Tetrahedron 2001; 57: 6329
- 86 Yu W, McConathy J, Olson J, Lager VM, Goodman MM. J Med Chem 2007; 50: 6718
- 87 Yu W, Williams L, Malveaux E, Camp VM, Olson J, Goodman MM. Bioorg Med Chem Lett 2008; 18: 1264
- 88 Bartels JL, Huang C, Li A. et al. J Med Chem 2015; 58: 8542
- 89 Fuchibe K, Hatemata R, Akiyama T. Tetrahedron 2006; 62: 11304
- 90 Thapaliya ER, Zhao J, Ellis-Davies GCR. ACS Chem Neurosci 2019; 10: 2481
