

RSS-Feed abonnieren
DOI: 10.1055/s-0037-1609517
Synthesis of Phthalimides: A New Entry via TBAI-Catalyzed Intramolecular Cyclization of 2-(Hydroxymethyl)benzamides
Autoren
K. N. and N. R. thank CSIR New Delhi for Research Fellowships. This research work was financially supported by the CSIR, New Delhi (BSC 0116).
Publikationsverlauf
Received: 21. März 2018
Accepted after revision: 19. April 2018
Publikationsdatum:
16. Mai 2018 (online)

Communication No. IICT/Pubs./2018/080
Abstract
Herein we report an unprecedented metal-free TBAI/TBHP mediated C–N bond formation via intramolecular cyclization of 2-(hydroxymethyl)benzamides to furnish N-substituted phthalimides in excellent yields.
Keywords
phthalimide - intramolecular cyclization - transition-metal free - C–N bond formation - oxidationPhthalimides are frequently encountered as core structures in natural products, pharmaceuticals and agrochemicals,[1] and phthalimde derivatives play a role in histone deacetylase (HDAC) inhibition.[2] Some selected examples are shown in Figure [1].
The traditional strategy used for the synthesis of phthalimides involves condensation of phthalic acids or anhydrides and primary amines in refluxing organic solvents. However, due to lengthy reaction times or the use of expensive auxiliary reagents, such methods are not entirely satisfactory. In recent years, several significant novel approaches that provide ready access to phthalimides have been reported. Hong and co-workers[3] used ruthenium in the presence of a ligand for the synthesis of cyclic imides from diols. Others have converted phthalic anhydrides into phthalimides under HMDS-Lewis acid conditions[4] or by using lanthanide oxides.[5] Amino alcohols have been converted into lactams under Ru-catalysis,[6] OSU-6 has been used to catalyze transamidation of acids and esters[7] and enamines and amines have been oxidatively coupled.[8]

With the aim of replacing transition-metal-assisted protocols, this communication describes our efforts towards the development of a new synthetic method using metal-free catalyst systems. To date, no procedures have been developed for the preparation of phthalimides via tetrabutylammonium iodide (TBAI)/tert-butyl hydrogen peroxide (TBHP)-mediated intramolecular oxidative cyclization of 2-hydroxymethylbenzamides. Herein, we report an alternative approach for the synthesis of substituted phthalimides starting from 2-hydroxymethylbenzamides.
We started our investigation into the TBAI-catalyzed intramolecular cyclization of 1aunder the following reaction conditions: TBAI (0.2 mmol), H2O2 (5 mmol) in tetrahydrofuran (THF) at 80 °C for 12 h, whereupon the desired 2-benzylisoindoline-1,3-dione 2a was obtained in 8% yield (Table [1], entry1). Encouraged by this result, we screened a variety of oxidants (entries 2 and 3), and found that the product yield could be improved to 43% when TBHP was used as the oxidant (entry 3). We then moved to solvent screening and found EtOAc at reflux to be the most efficient solvent for this transformation, affording 2a in 96% yield (entry 4). Furthermore, the same reaction under optimized reaction conditions but at room temperature gave only trace amounts of 2a (entry 5).
Various catalysts were then screened (Table [1], entries 6–8). As expected, in the absence of catalyst and oxidant (TBAI/TBHP in EtOAc) no product was observed (entries 9 and 10); whereas reaction without added oxidant but open to the atmosphere furnished 24% yield of the product (entry 11). The synthetic protocol disclosed herein involves C(sp3)–N bond formation, and N-substituted phthalimides can be synthesized in good yields from 2-(hydroxymethyl)benzamides.
a Reaction conditions: 1a (1.0 mmol), catalyst (0.2 mmol), oxidant (5.0 mmol), solvent (2 mL), 8–24 h under N2 atmosphere.
b Isolated yield.
After establishing the optimized reaction conditions, next we explored the substrate scope of this reaction (Scheme [1]). All the required substrates were prepared by using a procedure from phthalides and amines in the presence of AlCl3 at room temperature to give the corresponding 2-(hydroxymethyl)benzamides 1a–q in good yields.[9]
Initially, we studied the effect of the substituent on the amine component. Substrates with electron-withdrawing or -donating groups such as Cl, OMe on the phenyl ring furnished the desired products 2b and 2c in 91% and 90% yield, respectively (Scheme [1]). N-Aliphatic linked substrates 1d, 1e, 1f, and 1i were also successful under these reaction conditions, affording the products 2d (78%), 2e (84%), 2f (92%), and 2i (67%), respectively. We also examined chiral substrates 1g and 1h, and found excellent conversion into 2g (92%) and 2h (85%), respectively, without any racemization.

Heteroatom containing thiophene 1k and pyridine 1l substitution were tolerated well, with the reaction conditions delivering the corresponding products 2k (81%) and 2l (87%), respectively. 2-(Hydroxymethyl)benzamides 1m–q also underwent facile oxidative cyclization to give the corresponding phthalmides in good yields (71–87%); the exception being 1q, which gave a moderate yield of 2q (52%).
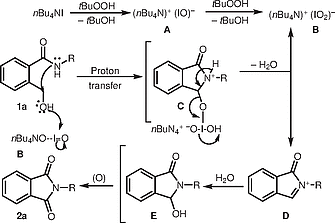
Based on the above results, a plausible mechanism for the TBAI/TBHP mediated oxidative cyclization of 1a is shown in Scheme [2]. On the basis of reported precedent,[10] the reaction is proposed to be initiated by TBAI, which, when reacted with TBHP, initially generates tetra-n-butyl ammonium hypoiodite A, which is further oxidized by TBHP to produce iodite complex B. Subsequently, intramolecular cyclization takes place at the benzylic position of 1a, mediated by iodite B, to afford intermediate C in which proton transfer from quaternary ammonium ion followed by dehydration gives rise to iminium ion D. The released hypoiodate A can be reoxidized by TBHP to regenerate B. Finally, addition of water and subsequent oxidation of D produces the desired product 2a.
In conclusion, we have demonstrated a simple oxidative cyclization to produce phthalimide derivatives. The method uses commercially available reagents TBAI and TBHP and the single-step protocol offers an atom-economical strategy. The precursor acyclic amides are readily accessible from benzoic acid derivatives.
TLC analysis was performed on Merck 60 F254 silica gel plates and the developed plates were visualized by exposure to ultraviolet light and/or α-naphthol charring. Organic extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure in a Büchi rotary evaporator. All column chromatographic separations were performed using silica gel (SiO2; 60–120 mesh) with EtOAc and hexane as eluents. 1H NMR spectra were recorded at 400 and 500 MHz (using TMS as reference), and 13C NMR were recorded at 100 and 125 MHz (using the CDCl3 triplet centered at δ = 77.0 Hz as reference) in CDCl3 as solvent at ambient temperature. Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), and coupling constants (Hz). Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are given in Hz. Mass spectrometry was performed in ESI mode. High-resolution mass spectra (HRMS) were obtained using either a TOF or a double focusing spectrometer. For low (MS) and high (HRMS) resolution, m/z ratios are reported as values in atomic mass units. Melting points were recorded with a Büchi 535 melting point apparatus and are uncorrected.
All chemicals were purchased from Sigma–Aldrich and S.D Fine Chemicals, Pvt. Ltd. India and used as received.
Synthesis of 2-Benzylisoindoline-1,3-diones; General Procedure
The requisite N-benzyl-2-(hydroxymethyl)benzamide (1.0 mmol) was dissolved in anhydrous EtOAc (2 mL). TBAI (0.2 mmol) and TBHP (5.0 mmol, 5 equiv) were added, and the mixture was stirred at 80 °C for 8 h and monitored by TLC. After completion of the reaction, the mixture was cooled to r.t., washed with water (10 mL), dried over anhydrous Na2SO4, filtered, evaporated under reduced pressure, and the residue was purified by column chromatography (hexane/EtOAc, 10:2) to furnish the product phthalimide.
2-Benzylisoindoline-1,3-dione (2a)[11]
Yield: 0.249 g (96%); white solid; m.p 108–110 °C; R f = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.85–7.83 (m, 2 H), 7.71–7.69 (m, 2 H), 7.44–7.42 (m, 2 H), 7.33–7.25 (m, 3 H), 4.85 (s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 168.0, 136.3, 134.0, 132.1, 128.7, 128.6, 127.8, 123.3, 41.6.
IR (neat): 3291, 3066, 2922, 2852, 1713, 1491, 1219, 1091, 1014, 772 cm–1.
2-(4-Methoxybenzyl)isoindoline-1,3-dione (2b)
Yield: 0.225 g (91%); white solid; m.p 120–122 °C; R f = 10:4 (hexane/EtOAc).
1H NMR (400 MHz, CDCl3): δ = 7.84–7.82 (m, 2 H), 7.70–7.68 (m, 2 H), 7.39–7.37 (m, 2 H), 6.85–6.82 (m, 2 H), 4.78 (s, 2 H), 3.77 (s, 3 H).
13C NMR (100 MHz, CDCl3): δ = 168.1, 159.2, 133.9, 132.1, 130.1, 128.6, 123.3, 114.0, 55.2, 41.0.
IR (neat): 3063, 2958, 2922, 2852, 1761, 1509, 1246, 933, 711, 575 cm–1.
HRMS (ESI): m/z [M + H] calcd for C16H14NO3: 268.0968; found: 268.0968.
2-(4-Chlorobenzyl)isoindoline-1,3-dione (2c)[12]
Yield: 0.223 g (90%); white solid; m.p 120–122 °C; R f = 10:3 (hexane/EtOAc).
1H NMR (400 MHz, CDCl3): δ = 7.85 (dd, J = 5.3, 3.0 Hz, 2 H), 7.72 (dd, J = 5.3, 3.0 Hz, 2 H), 7.39–7.36 (m, 2 H), 7.28 (dt, J = 5.1, 2.8 Hz, 2 H), 4.81 (s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 167.9, 134.8, 134.2, 133.8, 132.0, 130.1, 128.8, 123.4, 40.9.
IR (neat): 3291, 3066, 2922, 2852, 1713, 1637, 1546, 1219, 1014, 772 cm–1.
2-Allylisoindoline-1,3-dione (2d)
Yield: 0.19 g (78%); white solid; m.p. 57–60 °C; R f = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.87–7.85 (m, 2 H), 7.74–7.71 (m, 2 H), 5.93–5.86 (m, 1 H), 5.27–5.18 (m, 2 H), 4.30 (dt, J = 5.7, 1.5 Hz, 2 H).
13C NMR (100 MHz, CDCl3): δ = 167.9, 133.9, 132.1, 131.5, 123.3, 117.7, 77.3, 77.0, 76.7, 40.0.
IR (neat): 3084, 3021, 2921, 2854, 1708, 1610, 1388, 1188, 846, 724 cm–1.
HRMS (ESI): m/z [M + H] calcd for C11H10NO2: 188.0706; found: 188.0705.
2-(Prop-2-yn-1-yl)isoindoline-1,3-dione (2e)[11]
Yield: 0.205 g (84%); white solid; m.p. 55–58 °C; R f = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.89 (dd, J = 5.4, 3.1 Hz, 2 H), 7.75 (dd, J = 5.5, 3.0 Hz, 2 H), 4.46 (d, J = 2.5 Hz, 2 H), 2.23 (d, J = 2.5 Hz, 1 H).
13C NMR (126 MHz, CDCl3): δ = 167.0, 134.2, 132.0, 123.6, 77.3, 77.2, 77.0, 76.8, 71.5, 27.
IR (neat): 3291, 3090, 2962, 2922, 2852, 1768, 1396, 1120, 923, 772 cm–1.
2-Cyclopropylisoindoline-1,3-dione (2f)
Yield: 0.225 g (92%); white solid; m.p. 78–83 °C; R f = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.83–7.81 (m, 2 H), 7.71 (dd, J = 5.5, 3.1 Hz, 2 H), 2.74–2.69 (m, 1 H), 1.04–0.99 (m, 4 H).
13C NMR (126 MHz, CDCl3): δ = 168.8, 133.9, 131.7, 123.1, 77.3, 77.0, 76.8, 30.9, 20.9, 5.21.
IR (neat): 3021, 2923, 2854, 1765, 1710, 1398, 1067, 818, 771, 715 cm–1.
HRMS (ESI): m/z [M + H] calcd for C11H10NO2: 188.0706; found: 188.0706.
(R)-2-(1-Phenylethyl)isoindoline-1,3-dione (2g)
Yield: 0.227 g (92%); oil; Rf = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.80–7.79 (m, 2 H), 7.69–7.65 (m, 2 H), 7.53–7.49 (m, 2 H), 7.36–7.31 (m, 2 H), 7.27–7.23 (m, 1 H), 5.57 (q, J = 7.3 Hz, 1 H), 1.93 (d, J = 7.3 Hz, 3 H).
13C NMR (126 MHz, CDCl3): δ = 168.1, 140.3, 133.9, 132.0, 128.5, 127.7, 127.4, 123.2, 49.6, 17.5.
IR (neat): 3270, 3062, 2853, 1631, 1449, 1276, 1011, 876, 771, 699 cm–1.
HRMS (ESI): m/z [M + H] calcd for C16H14NO2: 252.1019; found: 252.1020.
(S)-2-(1-Phenylethyl)isoindoline-1,3-dione (2h)
Yield: 0.210 g (85%); R f = 10:2 (hexane/EtOAc); oil.
1H NMR (400 MHz, CDCl3): δ = 7.79 (dd, J = 5.5, 2.9 Hz, 2 H), 7.66 (dt, J = 7.7, 3.8 Hz, 2 H), 7.51 (d, J = 7.4 Hz, 2 H), 7.32 (t, J = 7.4 Hz, 2 H), 7.25 (t, J = 7.3 Hz, 2 H), 5.56 (t, J = 7.3 Hz, 1 H), 1.93 (d, J = 7.3 Hz, 3 H).
13C NMR (126 MHz, CDCl3): δ = 168.1, 141.6, 133.9, 132.0, 128.5, 127.7, 127.4, 123.1, 49.6, 17.6.
IR (neat): 3271, 3130, 3011, 1710, 1445, 1276, 1011, 876, 771, 699 cm–1.
HRMS (ESI): m/z [M + H] calcd for C16H14NO2: 252.1019; found: 252.1020.
2-Butylisoindoline-1,3-dione (2i)
Yield: 0.165 g (67%); oil; R f = 10:3 (hexane/EtOAc).
1H NMR (400 MHz, CDCl3): δ = 7.82 (dt, J = 6.9, 3.5 Hz, 2 H), 7.74–7.69 (m, 2 H), 3.68 (t, J = 7.3 Hz, 2 H), 1.71–1.62 (m, 2 H), 1.36 (dt, J = 14.8, 7.4 Hz, 2 H), 0.95 (t, J = 7.4 Hz, 3 H).
13C NMR (126 MHz, CDCl3): δ = 168.3, 133.7, 132.1, 123.0, 37.6, 30.5, 20.0, 13.5.
IR (neat): 2958, 2870, 1771, 1437, 1363, 1051, 940, 772, 717, 618 cm–1.
HRMS (ESI): m/z [M + H] calcd for C12H14NO2: 204.1021; found: 204.1022.
5-Fluoro-2-(3,4,5-trimethoxybenzyl)isoindoline-1,3-dione (2j)
Yield: 0.217 g (88%); white solid; m.p. 138–142 °C; R f = 10:5 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.85 (dd, J = 8.2, 45 Hz, 1 H), 7.53–7.51 (m, 1 H), 7.40–7.35 (m, 1 H), 6.69 (s, 2 H), 4.75 (s, 2 H), 3.85 (s, 6 H), 3.80 (s, 3 H).
13C NMR (126 MHz, CDCl3): δ = 166.9, 165.3, 153.3, 137.7, 134.9, 131.7, 127.8, 125.7, 123.1, 121.1, 111.3, 111.1, 106.0, 60.7, 56.1, 42.1.
IR (neat): 3063, 2958, 2922, 2852, 1756, 1717, 1246, 933, 711, 680 cm–1.
HRMS (ESI): m/z [M + H] calcd for C18H17FNO5: 346.1085; found: 346.1092.
2-(Thiophene-2-ylmethy)isoindoline-1,3-dione (2k)
Yield: 0.2 g (81%); white solid; m.p. 114–117 °C; R f = 10:3 (hexane/EtOAc).
1H NMR (400 MHz, CDCl3): δ = 7.86–7.83 (m, 2 H), 7.72–7.69 (m, 2 H), 7.21 (dd, J = 5.1, 1.2 Hz, 1 H), 7.15–7.14 (m, 1 H), 6.93 (dd, J = 5.1, 3.5 Hz, 1 H), 5.02 (s, 2 H).
13C NMR (126 MHz, CDCl3): δ = 167.5, 138.0, 134.0, 132.0, 127.7, 126.9, 125.9, 123.4, 35.7.
IR (neat): 3057, 2964, 2841, 1740, 1723, 1355, 1285, 1240, 709, 666 cm–1.
HRMS (ESI): m/z [M + H] calcd for C13H10NO2S: 244.1023; found: 244.1025.
2-(Pyridin-2-ylmethyl)isoindoline-1,3-dione (2l)
Yield: 0.215 g (87%); white solid; m.p. 121–124 °C; R f = 10:5 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 8.53–8.51 (m, 1 H), 7.88 (dd, J = 5.4, 3.1 Hz, 2 H), 7.73 (dd, J = 5.5, 3.0 Hz, 2 H), 7.65–7.62 (m, 1 H), 7.28 (d, J = 7.4 Hz, 1 H), 7.16 (dd, J = 7.2, 5.0 Hz, 2 H), 5.02 (s, 2 H).
13C NMR (100 MHz, CDCl3): δ = 168.1, 155.3, 149.6, 136.7, 134.0, 132.2, 123.5, 122.5, 121.5, 76.7, 42.9.
IR (neat): 3029, 1784, 1709, 1612, 1482, 1430, 1387, 1109, 794, 722 cm–1.
HRMS (ESI): m/z [M + H] calcd for C14H11N2O2: 239.0815; found: 239.0814.
2-Phenylisoindoline-1,3-dione (2m)
Yield: 0.215 g (87%); white solid; m.p. 93–102 °C; R f = 10:2 (hexane/EtOAc).
1H NMR (400 MHz, CDCl3): δ = 7.97 (dd, J = 5.5, 3.0 Hz, 2 H), 7.80 (dd, J = 5.5, 3.1 Hz, 2 H), 7.54–7.49 (m, 2 H), 7.46–7.39 (m, 3 H).
13C NMR (100 MHz, CDCl3): δ = 167.3, 134.4, 131.8, 131.7, 129.1, 128.1, 126.6, 123.7.
IR (neat): 3076, 1709, 1595, 1496, 1388, 1117, 881, 761, 718 cm–1.
HRMS (ESI): m/z [M + H] calcd for C14H10NO2: 224.0706; found: 224.0707.
2-(4-Bromophenyl)isoindoline-1,3-dione (2n)[11]
Yield: 0.175 g (71%); white solid; m.p. 118–123 °C; R f = 10: 2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.96 (dd, J = 5.4, 3.1 Hz, 2 H), 7.81 (dd, J = 5.4, 3.0 Hz, 2 H), 7.64 (dd, J = 5.4, 3.0 Hz, 2 H), 7.36 (d, J = 8.7 Hz, 2 H).
13C NMR (126 MHz, CDCl3): δ = 166.9, 134.6, 132.3, 131.6, 130.7, 127.9, 123.9, 121.8.
IR (neat): 3065, 2957, 2921, 1715, 1463, 1124, 1024, 818, 772, 714 cm–1.
2-(3-Bromophenyl)isoindoline-1,3-dione (2o)
Yield: 0.198 g (80%); white solid; m.p. 118–125 °C; R f = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.97 (dd, J = 5.4, 3.1 Hz, 2 H), 7.81 (dd, J = 5.5, 3.0 Hz, 2 H), 7.65 (t, J = 1.8 Hz, 1 H), 7.55–7.53 (m, 1 H), 7.45–7.36 (m, 2 H).
13C NMR (100 MHz, CDCl3): δ = 166.8, 134.6, 131.5, 131.1, 130.3, 129.5, 125.1, 123.9, 122.4.
IR: 3062, 2986, 2805, 1715, 1463, 1265, 987, 816, 772, 716 cm–1.
HRMS (ESI): m/z [M + H] calcd for C14H9BrNO2: 301.1132; found: 301.1135.
2-(3-Fluorophenyl)isoindoline-1,3-dione (2p)
Yield: 0.19 g (77%); white solid; m.p. 113–115 °C; R f = 10: 2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.98–7.96 (m, 2 H), 7.81 (dd, J = 5.5, 3.0 Hz, 2 H), 7.50–7.45 (m, 1 H), 7.29 (dd, J = 8.1, 1.8 Hz, 1 H), 7.26–7.23 (m, 1 H), 7.14–7.23 (m, 1 H).
13C NMR (125 MHz, CDCl3): δ = 166.8, 163.6, 161.6, 134.6, 133.1, 133.0, 131.5, 130.2, 123.9, 122.0, 115.1, 114.9, 114.0, 113.8.
IR (neat): 3294, 2949, 2837, 1646, 1450, 1413, 1219, 1113, 1014, 772 cm–1.
HRMS (ESI): m/z [M + H] calcd for C14H9FNO2: 242.0210; found: 242.0213.
2-(3,5-Difluorophenyl)isoindoline-1,3-dione (2q)
Yield: 0.128 g (52%); white solid; m.p. 132–135 °C; Rf = 10:2 (hexane/EtOAc).
1H NMR (500 MHz, CDCl3): δ = 7.99–7.97 (m, 2 H), 7.84–7.82 (m, 2 H), 7.45–7.44 (m, 2 H), 7.40 (t, J = 1.8 Hz, 1 H).
13C NMR (126 MHz, CDCl3): δ = 166.4, 135.2, 134.8, 133.5, 131.3, 128.1, 124.8.
IR (neat): 3091, 2922, 2853, 1727, 1447, 1220, 1079, 806, 772, 665 cm–1.
HRMS (ESI): m/z [M + H] calcd for C14H8Cl2NO2: 292.1200; found: 292.1205.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/s-0037-1609517.
- Supporting Information (PDF) (opens in new window)
-
References
- 1a Matsumoto K. Nagashima K. Kamigauchi T. Kawamura Y. Yasuda Y. Ishii K. Uotani N. Sato T. Nakai H. Terui Y. Kikuchi J. Ikenisi Y. Yoshida T. Kato T. Itazaki H. J. Antibiot. 1995; 4: 439
- 1b Miyachi H. Azuma A. Ogasawara A. Uchimura E. Watanabe N. Kobayashi Y. Kato F. Kato M. Hashimoto Y. J. Med. Chem. 1997; 40: 2858
- 1c Figg WD. Raje S. Bauer KS. Tompkins A. Venzon D. Bergan R. Chen A. Hamilton M. Pluda J. Reed E. J. Pharm. Sci. 1999; 88: 121
- 1d Balzarini EJ. Clercq D. Kaminska B. Orzeszko A. Antiviral Chem. Chemother. 2003; 14: 139
- 1e Franks ME. Macpherson GR. Figg WD. Lancet 2004; 363: 1802
- 1f Luzzio FA. Duveau DY. Lepper ER. Figg WD. J. Org. Chem. 2005; 70: 10117
- 1g Shoji A. Kuwahara M. Ozaki H. Sawai H. J. Am. Chem. Soc. 2007; 129: 1456
- 2 Shinji C. Maeda S. Imai K. Yoshida M. Hashimoto Y. Miyachi H. Bioorg. Med. Chem. 2006; 14: 7625
- 3 Zhang J. Senthilkumar M. Ghosh SC. Hong SH. Angew. Chem. Int. Ed. 2010; 49: 6391
- 4 Reddy PY. Kondo S. Toru T. Ueno Y. J. Med. Chem. 1997; 62: 2652
- 5 Ali MA. Moromi SK. Touchy AS. Shimizu KI. ChemCatChem 2016; 8: 891
- 6 Naota T. Murahashi SI. Synlett 1991; 693
- 7 Nammalwar B. Muddala NP. Watts FM. Bunce RA. Tetrahedron 2015; 9101
- 8a Hall A. Billinton A. Bristow AK. Brown SH. Chowdhury A. Cutler L. Giblin MP. G. Hayhow TG. Kilford IR. Naylor A. Passingham B. Rawlings DA. Bioorg. Med. Chem. Lett. 2008; 18: 4027
- 8b Yuan Y. Hou W. Negrerie DZ. Zhao K. Du Y. Org. Lett. 2014; 16: 5410
- 8c Aruri H. Singh U. Kumar S. Kushwaha M. Gupta AP. Vishwakarma RA. Singh PP. Org. Lett. 2016; 18: 3638
- 9 Jin Y. Fu H. Yin Y. Jiang Y. Zhaoa Y. Synlett 2007; 901
- 10 Yuan Y. Hou W. Negrerie DZ. Zhao K. Du Y. Org. Lett. 2014; 16: 5410
- 11 Rohith S. Rao AS. Pralhad JN. Vishal R. Chem. Commun. 2015; 473
- 12 Nammalwar B. Muddala NP. Watts FM. Bunce RA. Tetrahedron 2015; 71: 9101
-
References
- 1a Matsumoto K. Nagashima K. Kamigauchi T. Kawamura Y. Yasuda Y. Ishii K. Uotani N. Sato T. Nakai H. Terui Y. Kikuchi J. Ikenisi Y. Yoshida T. Kato T. Itazaki H. J. Antibiot. 1995; 4: 439
- 1b Miyachi H. Azuma A. Ogasawara A. Uchimura E. Watanabe N. Kobayashi Y. Kato F. Kato M. Hashimoto Y. J. Med. Chem. 1997; 40: 2858
- 1c Figg WD. Raje S. Bauer KS. Tompkins A. Venzon D. Bergan R. Chen A. Hamilton M. Pluda J. Reed E. J. Pharm. Sci. 1999; 88: 121
- 1d Balzarini EJ. Clercq D. Kaminska B. Orzeszko A. Antiviral Chem. Chemother. 2003; 14: 139
- 1e Franks ME. Macpherson GR. Figg WD. Lancet 2004; 363: 1802
- 1f Luzzio FA. Duveau DY. Lepper ER. Figg WD. J. Org. Chem. 2005; 70: 10117
- 1g Shoji A. Kuwahara M. Ozaki H. Sawai H. J. Am. Chem. Soc. 2007; 129: 1456
- 2 Shinji C. Maeda S. Imai K. Yoshida M. Hashimoto Y. Miyachi H. Bioorg. Med. Chem. 2006; 14: 7625
- 3 Zhang J. Senthilkumar M. Ghosh SC. Hong SH. Angew. Chem. Int. Ed. 2010; 49: 6391
- 4 Reddy PY. Kondo S. Toru T. Ueno Y. J. Med. Chem. 1997; 62: 2652
- 5 Ali MA. Moromi SK. Touchy AS. Shimizu KI. ChemCatChem 2016; 8: 891
- 6 Naota T. Murahashi SI. Synlett 1991; 693
- 7 Nammalwar B. Muddala NP. Watts FM. Bunce RA. Tetrahedron 2015; 9101
- 8a Hall A. Billinton A. Bristow AK. Brown SH. Chowdhury A. Cutler L. Giblin MP. G. Hayhow TG. Kilford IR. Naylor A. Passingham B. Rawlings DA. Bioorg. Med. Chem. Lett. 2008; 18: 4027
- 8b Yuan Y. Hou W. Negrerie DZ. Zhao K. Du Y. Org. Lett. 2014; 16: 5410
- 8c Aruri H. Singh U. Kumar S. Kushwaha M. Gupta AP. Vishwakarma RA. Singh PP. Org. Lett. 2016; 18: 3638
- 9 Jin Y. Fu H. Yin Y. Jiang Y. Zhaoa Y. Synlett 2007; 901
- 10 Yuan Y. Hou W. Negrerie DZ. Zhao K. Du Y. Org. Lett. 2014; 16: 5410
- 11 Rohith S. Rao AS. Pralhad JN. Vishal R. Chem. Commun. 2015; 473
- 12 Nammalwar B. Muddala NP. Watts FM. Bunce RA. Tetrahedron 2015; 71: 9101
