
RSS-Feed abonnieren
DOI: 10.1055/s-0042-1758147
Anti–Gamma Aminobutyric Acid B Autoimmune Encephalitis in an Indian Child with Early-Onset Seizures, Neurodegeneration, and Brain Calcification due to NRROS Variation: The First Reported Case Worldwide

Abstract
A 1.5-year-old boy presented to us with a history of normal growth and developmental parameters until 6 months of age. However, at 7 months of age, he developed multiple types of seizures consisting initially of complex febrile seizures, followed by afebrile seizures. Multifocal clonic, generalized tonic–clonic, and myoclonic (multifocal and generalized) were the evolving seizure types. He had truncal hypotonia, but his appendicular hypotonia progressed to hypertonia over the next few months and further to decorticate posturing. Brain magnetic resonance imaging (MRI) showed generalized atrophy, predominantly frontotemporal, without any focal signal abnormalities or contrast enhancement. Computed tomography (CT) showed speckled calcification in subcortical white matter. Electroencephalogram showed bilateral frontotemporal epileptiform discharges with secondary generalization. His cerebrospinal fluid had normal cytology and biochemical results but was positive for anti–gamma aminobutyric acid B antibodies. Whole exome sequencing showed likely pathogenic, novel autosomal recessive homozygous variation of NRROS gene on chromosome 3 [c.1487G > A (p.Trp496Ter)], which impairs the functioning of anti-inflammatory cytokine transforming growth factor beta, resulting in a proinflammatory state within the central nervous system and thereby promoting autoimmune encephalitis. Parental Sanger sequencing validated the variation in both his parents. He was treated with both pulse methylprednisolone (30 mg/kg/day for 5 days) and intravenous immunoglobulin (2 g/kg), followed by slowly tapering of oral prednisolone and monthly intravenous immunoglobulin infusion (1 g/kg). There was significant reduction in seizure frequency and disappearance of epileptiform discharges from the electroencephalogram. However, the motor and cognitive improvement did not occur, and he had microcephaly and growth failure at the last follow-up. This is the 11th case report of neurodegeneration associated with NRROS gene variations, but the first report of autoimmune encephalitis being triggered by the variation in a child.
#
Introduction
Microglia, the long-living innate immune cells in the brain, are hosts to innumerable genes, which encode for a plethora of central nervous system (CNS) disorders—neurodevelopmental, neurodegenerative, neuroinflammatory, and neoplastic.[1] CSF1R, DAP12, TYROBP/TREM2, USP18, IRF8, and NRROS genes have been linked to diverse phenotypes of microgliopathies.[2] NRROS (negative regulator of reactive oxygen species) is a transmembrane protein of the endoplasmic reticulum containing leucine-rich repeats, whose function was first described by Noubade et al in 2014.[3] Phagocytes produce reactive oxygen species (ROS) to eliminate invading pathogens. However, tight regulation is needed during inflammatory processes to prevent collateral tissue damage. NRROS controls NADPH oxidase 2 (NOX2) degradation and toll-like receptor signaling and activates the latent anti-inflammatory cytokine transforming growth factor beta (TGF-β) by anchoring it over the surfaces of macrophages and microglia.[4] Thus, NRROS regulates ROS generation, thereby minimizing collateral tissue damage. In mice experiments, NRROS-deficient phagocytes exhibited elevated ROS production during inflammation, with resultant enhanced microbiocidal activity. However, due to oxidative CNS tissue damage, these mice developed fatal autoimmune encephalomyelitis.[4] [5]
Loss-of-function variants in the NRROS gene result in infantile-onset encephalopathy characterized by early-onset seizures with neurodegeneration and brain calcification (SENEBAC), publications on which are exceedingly rare. Two articles[2] [4] describing nine cases in 2020 were followed by a recent case report in April 2022.[6] Ours is the 11th case reported worldwide and the first from India. Moreover, it is the first report to describe associated autoimmune encephalitis in a child with a NRROS variation.
#
Case Report
The index case is of a second-born boy from a second-degree consanguineous marriage. The boy had normal growth and developmental milestones until 6 months. At 7 months, he had his first episode of febrile status epilepticus, followed by recurrent episodes of complex febrile seizures. At 9 months, he had an afebrile generalized tonic–clonic seizure, soon followed by the appearance of myoclonic jerks, which were both multifocal and generalized, and occurred in clusters of five to seven jerks in each cluster. He also had clonic jerks involving both upper limbs. His multiple seizure types progressed in frequency and severity, associated with global developmental regression. His family history was noncontributory to any type of epilepsy.
He presented to our center at 1.5 years of age. There were no dysmorphisms, neurocutaneous markers, abnormal body or urinary odors, or organomegaly. His hospital discharge reports after birth showed normal birth weight and head circumference. He initially had both axial and appendicular hypotonia, followed by the appearance of appendicular hypertonia over the next 3 months, brisk deep tendon reflexes, and bilateral extensor plantar responses. Subsequently, he developed decorticate posturing (clinical photographs in [Supplemental File 1], available in online version only). He was treated with multiple antiepileptic drugs (AEDs) such as levetiracetam, clobazam, and valproate, but without any significant benefit.
|
Author |
Genetic variations |
Type of variation |
Zygosity |
|---|---|---|---|
|
Smith et al[2] |
c.1777C > T/p.(Gln593*) |
Nonsense |
Homozygous |
|
c.1257del/p.(Gly420AlafsTer14) |
Nonsense |
Homozygous |
|
|
Dong et al[4] |
c.1981delC, p.Leu661Serfs∗97 |
Frameshift |
Homozygous |
|
c.1644delG, p.Thr549Profs∗82 |
Frameshift |
Homozygous |
|
|
variant 1: c.190delC, p.Leu64Trpfs∗81 and variant 2: c.29T > C |
Frameshift |
Double heterozygous |
|
|
p.Leu10Pro |
Missense |
||
|
Macintosh et al[6] |
c.185T > C, p.Leu62Pro |
Missense |
Double heterozygous |
|
c.310C > T, p.Gln104Ter |
Nonsense |
||
|
Index case |
c.1487G > A (p.Trp496Ter) |
Nonsense |
Homozygous |
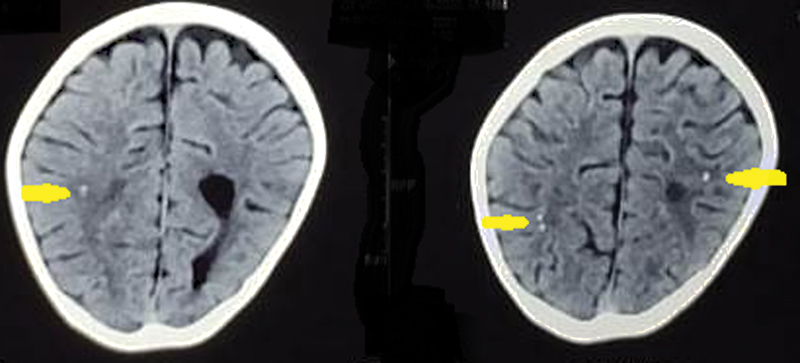
The complete hemogram, liver, renal, and thyroid function tests, cerebrospinal fluid (CSF) cell count, glucose and protein levels, and plasma lactate and ammonia levels were normal. The CSF autoimmune panel by indirect immunofluorescence test was positive for anti–gamma aminobutyric acid (anti-GABA) B antibody. Noncontrast computed tomography (CT) showed diffuse brain atrophy with punctuated scattered calcifications, predominantly in the subcortical white matter ([Fig. 1]). Magnetic resonance imaging (MRI) of the brain showed diffuse cerebral cortical and subcortical atrophy with relative sparing of the cerebellum and brainstem without any focal signal abnormalities or contrast enhancement ([Fig. 2]). The frontotemporal atrophy was more prominent than the parieto-occipital atrophy ([Fig. 3]). The blood spot tandem mass spectroscopy and urinary gas chromatography mass spectrometry were unremarkable for any metabolic disease. Electroencephalogram (EEG) showed predominantly bilateral frontocentrotemporal high-amplitude spike and sharp wave discharges ([Fig. 4]), with secondary generalization ([Fig. 5]), indicating encephalopathy. No sleep elements were seen during EEG. With a normal chest skiagram and abdominal ultrasound, other internal organ malformations were excluded. After obtaining parental consent, the next-generation sequencing of the whole exome was performed ([Supplemental File 1]) to look for genetic etiology.




Viewing the rapidly progressive encephalopathy in the background of anti-GABA B antibody positivity, we treated the child with pulse intravenous methylprednisolone (30 mg/kg/day for 5 days) and intravenous immunoglobulin (IVIG; 2 g/kg). Thereafter, his seizure frequency significantly reduced over the next 1 month. He was discharged on tapering doses of oral prednisolone, sodium valproate (60 mg/kg/day), levetiracetam (60 mg/kg/day), and clonazepam and was planned for monthly IVIG (1 g/kg) regime. The exome sequencing revealed a novel likely pathogenic autosomal recessive homozygous biallelic variation [c.1487G > A (p.Trp496Ter)] located on exon 3 of the NRROS gene. Sanger sequencing validated the presence of the variations in heterozygous state in both the parents ([Supplemental File 1], available in online version only).
He has been followed up over the last 8 months. He is currently on his prior AED regimen and oral prednisolone taper (currently 0.5 mg/kg/day) and has received 7 monthly IVIG pulses. Repeat CSF autoimmune profile, done 3 and 6 months after discharge, was negative for anti-GABA B antibody, but MRI brain showed mild progression of atrophic changes. He was seizure-free but significantly disabled with clinical decortication symptoms, without any speech output or visual tracking. His EEG showed significant improvement, without any epileptiform discharges, but still lacked the normal sleep elements ([Fig. 6]). At the last visit, his occipitofrontal circumference was 44 cm and his weight was 8.2 kg (both z-scores < − 3 of the World Health Organization growth charts).

#
Discussion
Smith et al described three children with SENEBAC (two girls and one boy) from two unrelated families, born out of nonconsanguineous marriage, in whom, after initial normal early development, the patients developed refractory seizures and neuroregression between 14 and 18 months, ultimately leading to death by the third year of life.[2] Histologically, there was widespread neuronal loss in the grey matter with reactive gliosis, neuropil calcification, and foamy macrophage infiltration in the perivascular spaces of the cerebral white matter from the frontal to the occipital cortices and cerebellar white matter.[2] Dong et al reported six children (three boys and 3 girls) from four unrelated families with seizure onset varying between 9 and 17 months.[4] Four children had mild-moderate developmental delay from the first year of life, while two had normal initial development with rapid regression after the onset of seizures. Multiple evolving seizure types were described: refractory focal seizures, infantile spasm syndrome, clonic, myoclonic, and complex partial seizures. Only one patient had onset of seizures along a febrile illness, similar to our child. Three children expired at 2 years 2 months, 3 years 3 months, and 4 years 2 months, whereas three children were alive at 20 months, 4 years 6 months, and 9 years 3 months when the article was published. Macintosh et al described the latest case who had ultrastructural defect in the mitochondria when examined by electron microscopy.[6] In their brain MRIs, these patients had corpus callosal thinning, delayed myelination, hypomyelination, and periventricular leukomalacia with diffuse cerebral and cerebellar white matter T2 hyperintensities, but subsequent images on follow-up revealed progressive severe generalized cortical and subcortical atrophy with resultant ex vacuo ventricular dilatation—similar to our case, whom we saw late in his disease course.
Microglia are categorized into M1 (classical) and M2 (alternative) phenotypes.[7] M1 phenotype promotes neurotoxicity by releasing proinflammatory cytokines (tumor necrosis factor alpha, interleukin 6 [IL-6], IL-1β, IL-12, ROS, nitric oxide, and expression of NOX2), while M2 phenotype induce neuroprotectivity by releasing anti-inflammatory cytokines (IL-10 and TGF-β).[8] ROS influence phenotypic switching of macrophages.[9] By αVβ8 integrin–dependent highly localized activation of TGF-β1 and downregulation of ROS and NOX2, NRROS suppress the M1 phenotype, hence promoting neuroprotection.[1] [2] [10] NRROS deficiency inactivates TGF-β1 due to loss of its anchorage over phagocytic cells. NRROS-deficient mice show presence of reactive microglia, and perivascular macrophages with elevated lysosomal content (indicator of increased phagocytic activity) populate the CNS, thereby promoting autoimmunity.[11] Oxidative stress due to ROS modifies and exposes the intraneuronal antigens to the immune system and results in neuroprogression, which is a progressive process of apoptosis, declining neurogenesis, reduced neuronal plasticity, and elevated autoimmune responses.[12]
None of the previously reported cases had associated autoimmune encephalitis triggered by NRROS variations. GABA-B encephalitis is common in the middle-aged and elderly males,[13] with only two pediatric cases reported till date,[14] [15] the youngest child being 3 years old. The clinical picture of refractory multiple seizure types with rapid global neuroregression signified severe encephalopathy in our case; hence, we chose combined first-line therapy with steroids and IVIG to treat his autoimmune encephalitis.[16] However, the extreme rarity of the entity in infancy and the absence of the characteristic MRI features of limbic encephalitis (temporal lobe, insular and hippocampal T2/fluid-attenuated inversion recovery hyperintense signal changes)[15] prompted us to screen for other (genetic) causes of rapid neuroregression. The resolution of the seizures and disappearance of the cerebral dysrhythmia with immunotherapy pointed toward the suppression of the CNS hyperimmune state promoted by the NRROS variation and the autoimmunity triggered by the genetic variation. IVIG induce microglial switch towards protective polarisation, and the ablation of these microglia led to the loss of neuroprotective effects of IVIG. Microglia stimulated by IVIG exhibited reduced neuronal apoptosis and death when exposed to glucose deprivation and ROS in vitro.[17] Although none of the previous authors have described the treatment modality, based on our experience with the index case and the above-mentioned experimental data, we propose immunomodulation as a possible approach to treat SENEBAC. Probably, earlier immunomodulation would have resulted in better functional outcome in our case. Viewing the CNS proinflammatory state in SENEBAC, immunomodulation would likely be effective in cases even without associated autoimmune encephalitis.
In our patient, the novel biallelic homozygous NM_198565.3(NRROS):c.1487G > A (p.Trp496Ter) variant is predicted to cause protein truncation at 496th amino acid instead of normal 692 amino acids with resultant >10% loss of normal protein function, and it has not been reported previously in gnomAD and 1kG. Sanger sequencing validated both the parents as the heterozygous carriers of the variation. [Table 1] gives a comprehensive account of the described variations of NRROS gene.[2] [4] [6]
#
What Does This Article Add?
-
First report of autoimmune encephalitis associated with SENEBAC, which was hitherto reported only in mouse experiments.
-
First to propose immunomodulation as a possible therapy for SENEBAC.
-
Adds another novel NRROS variation to the recently evolving genetic conundrum of SENEBAC.
#
#
Conflict of Interest
None declared.
-
References
- 1 Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol 2021; 39: 251-277
- 2 Smith C, McColl BW, Patir A. et al. Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol 2020; 139 (05) 947-951
- 3 Noubade R, Wong K, Ota N. et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 2014; 509 (7499): 235-239
- 4 Dong X, Tan NB, Howell KB. et al. Bi-allelic LoF-NRROS variants impairing active TGF-beta-1 delivery cause a severe infantile-onset neurodegenerative condition with intracranial calcification. Am J Hum Genet 2020; 106 (04) 559-569
- 5 Leavy O. Inflammation: regulating ROS. Nat Rev Immunol 2014; 14 (06) 357
- 6 Macintosh J, Derksen A, Poulin C. et al. Novel biallelic variants in NRROS associated with a lethal microgliopathy, brain calcifications, and neurodegeneration. Neurogenetics 2022; 23 (02) 151-156
- 7 Zhang J, Buller BA, Zhang ZG. et al. Exosomes derived from bone marrow mesenchymal stromal cells promote remyelination and reduce neuroinflammation in the demyelinating central nervous system. Exp Neurol 2022; 347: 113895
- 8 Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 2017; 35: 441-468
- 9 Nguyen HM, Grössinger EM, Horiuchi M. et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017; 65 (01) 106-121
- 10 Qin Y, Garrison BS, Ma W. et al. A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 2018; 174 (01) 156-171.e16
- 11 Du RH, Sun HB, Hu ZL, Lu M, Ding JH, Hu G. Kir6.1/K-ATP channel modulates microglia phenotypes: implication in Parkinson's disease. Cell Death Dis 2018; 9 (03) 404
- 12 Bakunina N, Pariante CM, Zunszain PA. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015; 144 (03) 365-373
- 13 Zhu F, Shan W, Lv R, Li Z, Wang Q. Clinical characteristics of anti-GABA-B receptor encephalitis. Front Neurol 2020; 11: 403
- 14 Premakumari NS, Rudrappa S, Patagar PM. Paediatric anti-GABAB receptor encephalitis associated with SARS-CoV-2 (COVID- 19) infection. Int J Contemp Pediatrics 2021; 8: 1909-1913
- 15 Kruer MC, Hoeftberger R, Lim KY. et al. Aggressive course in encephalitis with opsoclonus, ataxia, chorea, and seizures: the first pediatric case of γ-aminobutyric acid type B receptor autoimmunity. JAMA Neurol 2014; 71 (05) 620-623
- 16 Abound H, Probasco JC, Irani S. et al. Autoimmune Encephalitis Alliance Clinicians Network. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry 2021; 92 (07) 757-768
- 17 Häußler V, Daehn T, Rissiek B. et al. Intravenous immunoglobulin (IVIg) induce a protective phenotype in microglia preventing neuronal cell death in ischaemic stroke. Neuromolecular Med 2020; 22 (01) 121-132
Address for correspondence
Publikationsverlauf
Eingereicht: 04. August 2022
Angenommen: 20. September 2022
Artikel online veröffentlicht:
09. November 2022
© 2022. Thieme. All rights reserved.
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol 2021; 39: 251-277
- 2 Smith C, McColl BW, Patir A. et al. Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol 2020; 139 (05) 947-951
- 3 Noubade R, Wong K, Ota N. et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 2014; 509 (7499): 235-239
- 4 Dong X, Tan NB, Howell KB. et al. Bi-allelic LoF-NRROS variants impairing active TGF-beta-1 delivery cause a severe infantile-onset neurodegenerative condition with intracranial calcification. Am J Hum Genet 2020; 106 (04) 559-569
- 5 Leavy O. Inflammation: regulating ROS. Nat Rev Immunol 2014; 14 (06) 357
- 6 Macintosh J, Derksen A, Poulin C. et al. Novel biallelic variants in NRROS associated with a lethal microgliopathy, brain calcifications, and neurodegeneration. Neurogenetics 2022; 23 (02) 151-156
- 7 Zhang J, Buller BA, Zhang ZG. et al. Exosomes derived from bone marrow mesenchymal stromal cells promote remyelination and reduce neuroinflammation in the demyelinating central nervous system. Exp Neurol 2022; 347: 113895
- 8 Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 2017; 35: 441-468
- 9 Nguyen HM, Grössinger EM, Horiuchi M. et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017; 65 (01) 106-121
- 10 Qin Y, Garrison BS, Ma W. et al. A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 2018; 174 (01) 156-171.e16
- 11 Du RH, Sun HB, Hu ZL, Lu M, Ding JH, Hu G. Kir6.1/K-ATP channel modulates microglia phenotypes: implication in Parkinson's disease. Cell Death Dis 2018; 9 (03) 404
- 12 Bakunina N, Pariante CM, Zunszain PA. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015; 144 (03) 365-373
- 13 Zhu F, Shan W, Lv R, Li Z, Wang Q. Clinical characteristics of anti-GABA-B receptor encephalitis. Front Neurol 2020; 11: 403
- 14 Premakumari NS, Rudrappa S, Patagar PM. Paediatric anti-GABAB receptor encephalitis associated with SARS-CoV-2 (COVID- 19) infection. Int J Contemp Pediatrics 2021; 8: 1909-1913
- 15 Kruer MC, Hoeftberger R, Lim KY. et al. Aggressive course in encephalitis with opsoclonus, ataxia, chorea, and seizures: the first pediatric case of γ-aminobutyric acid type B receptor autoimmunity. JAMA Neurol 2014; 71 (05) 620-623
- 16 Abound H, Probasco JC, Irani S. et al. Autoimmune Encephalitis Alliance Clinicians Network. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry 2021; 92 (07) 757-768
- 17 Häußler V, Daehn T, Rissiek B. et al. Intravenous immunoglobulin (IVIg) induce a protective phenotype in microglia preventing neuronal cell death in ischaemic stroke. Neuromolecular Med 2020; 22 (01) 121-132