

RSS-Feed abonnieren
DOI: 10.1055/s-0033-1340162
A Phosphine-Free Approach to Primary Amides by Palladium-Catalyzed Aminocarbonylation of Aryl and Heteroaryl Iodides Using Methoxylamine Hydrochloride as an Ammonia Equivalent
Authors
Publikationsverlauf
Received: 29. August 2013
Accepted after revision: 30. September 2013
Publikationsdatum:
05. November 2013 (online)

Abstract
The palladium-catalyzed synthesis of primary amides by aminocarbonylation of aryl and heteroaryl iodides under phosphine-free conditions is reported for the first time. Methoxylamine hydrochloride, acting as an ammonia equivalent, undergoes sequential carbonylation and demethoxylation under mild reaction conditions. The procedure does not require a phosphine ligand and takes place in short reaction times at low temperatures to provide the products in excellent yields.
Primary aromatic and heteroaromatic amides constitute important classes of compounds due to their inherent bioactivity and the occurrence of this functional group in a plethora of biologically important compounds.[1] Primary amides act as synthetic platforms for further functionalization into primary amines by reduction,[2] or into nitriles by dehydration.[3] Traditionally, primary amides are synthesized by the reaction of activated carboxylic acid derivatives such as acid chlorides, anhydrides and esters with amines, hydration of the corresponding benzonitriles,[4] or by oxidation of benzylamines, benzyl alcohols, or benzaldehydes.[5] In addition, carbonylation chemistry can also provide an alternative route for the synthesis of aromatic primary amides. Compared to secondary and tertiary amide synthesis, the preparation of aromatic primary amides using carbonylation chemistry has not received similar attention.[1]
Various catalysts and phosphine ligands have been employed for carbonylation, along with different ammonia equivalents as nucleophilic partners. Such protocols have used palladium catalysts–ammonia equivalent combinations, such as [PdCl2(PPh3)4] and hexamethyldisilazane (HMDS),[6] and [PdCl2(PPh3)2] or [Pd(OAc)2(dppf)] and formamide.[7] Beller and co-workers used gaseous ammonia and Pd(OAc)2/dppf or Pd(OAc)2/nBuP(1-adamantyl)2 as the catalytic system for the synthesis of primary aromatic amides.[8] Mori et al. employed Pd2(dba)3/dppf as a catalytic system with titanium–nitrogen complexes as the amine source.[9] Skrydstrup reported the synthesis of primary aromatic amides using ammonium carbamate as an ammonia source and Pd(dba)2/JosiPhos as the catalytic system.[10a] In 2006, Larhed and co-workers used hydroxylamine hydrochloride as an ammonia equivalent along with Herrmann’s palladacycle and [(t-Bu)3PH]BF4 as an additional ligand, and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and N,N-diisopropylethylamine (DIPEA) as bases.[10b] Furthermore, Xu and Alper used Pd(OAc)2/CYTOP®292 along with aqueous ammonia for the aminocarbonylation of aryl iodides.[10c] However, the principal drawbacks of all these reported methods are the low stabilities, poor availability, and cost of the palladium complexes and phosphine ligands. Phosphine-free cross-coupling reactions have been investigated because of their reduced toxicity and cost burden, and the easier isolation of the product from the reaction mixture.[11]
Based on our research interest in phosphine-free carbonylation reactions,[12] we herein report an efficient palladium-catalyzed, phosphine-free protocol for the synthesis of aromatic primary amides using methoxylamine hydrochloride as an ammonia equivalent (Scheme [1]). Key to this success was the use of a palladium(II) chloride–sodium iodide (PdCl2–NaI) catalytic system in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) as the base. This protocol tolerates a wide range of functional groups and is applicable to aromatic and heteroaromatic substrates, providing excellent yields of products under mild reaction conditions.

Initially, we screened various palladium precursors using iodobenzene as the model substrate for the optimization of the reaction conditions (Table [1]). Palladium(II) chloride was found to be an effective catalyst in the presence of an iodide such as sodium iodide, potassium iodide or tetrabutylammonium iodide. In the absence of phosphine ligands, rapid deactivation of the catalyst takes place, but the iodide ligand was found to be effective for maintaining catalytic activity. The iodide can participate in several different steps in reactions catalyzed by transition metals, for example, during oxidative addition, migration, coupling or the reductive elimination steps.[13]
Sodium iodide was chosen as an effective iodide source for the carbonylation procedure (Table [1], entry 4). The reaction never proceeded to completion without the ligand source. Typically, catalytic experiments were performed at a low pressure of carbon monoxide (CO) (5 atm) at 90 °C. A polar solvent such as acetonitrile was found to be best, providing the desired products in eight hours (Table [1], entries 4–6). During our investigations on the influence of different bases, we discovered that 1,4-diazabicyclo[2.2.2]octane gave excellent results (Table [1], entries 4 and 9–11). It has been shown that N-methoxy-N-methylamide (Weinreb’s amide), under highly basic conditions, can demethoxylate to release formaldehyde as a significant side reaction (Scheme [2]).[14]
a Reaction conditions: iodobenzene (1 mmol), methoxylamine hydrochloride (1.2 equiv), PdCl2 (10 mol%), NaI (0.2 mmol), base (2 equiv), CO (5 atm), MeCN (15 mL), 90 °C, 8 h.
b Yields determined by GC.
c Reaction carried out using 5 mol% of PdCl2.

Using 1.2 equivalents of both 1,4-diazabicyclo[2.2.2]octane and methoxylamine hydrochloride, we observed formation of benzamide along with N-methoxybenzamide as an intermediate. Increasing the concentration of the base to two equivalents gave an excellent yield of benzamide. Hence, the methoxylamine hydrochloride underwent sequential carbonylation and demethoxylation.
An excess of 1,4-diazabicyclo[2.2.2]octane was used in all attempts, both to release free methoxylamine and for demethoxylation of the N-methoxybenzamide intermediate. With optimized parameters in hand, we next screened various aryl and heteroaryl iodide derivatives as substrates for aminocarbonylation.[15] Substrates with electron-donating (Me, OMe) and electron-withdrawing (NO2) substituents at ortho- and para-positions were successfully transformed into their corresponding primary amides in high yields (Table [2], entries 1–6). 1-Iodonaphthalene provided a 90% yield of the desired amide product (Table [2], entry 7). In the presence of other halogen substituents (F, Cl, Br), the aryl iodides were transformed selectively into the corresponding amides (Table [2], entries 8–11). Several heteroaryl iodides also provided excellent yields of the desired products (Table [2], entries 12–14). The analytical data of all the products were in accord with those reported in the literature.[8a] , [16a] [b] [c] [d]
|
Entry |
Aryl halide |
Product |
Yield (%)b |
|
1 |
 |
 |
96 |
|
2 |
 |
 |
93 |
|
3 |
 |
 |
96 |
|
4 |
 |
 |
89 |
|
5 |
 |
 |
93 |
|
6 |
 |
 |
87 |
|
7 |
 |
 |
90 |
|
8 |
 |
 |
94 |
|
9 |
 |
 |
85 |
|
10 |
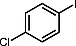 |
 |
91 |
|
11 |
 |
 |
84 |
|
12 |
 |
 |
92 |
|
13 |
 |
 |
88 |
|
14 |
 |
 |
86 |
a Reaction conditions: aryl iodide (1 mmol), methoxylamine hydrochloride (1.2 equiv), PdCl2 (10 mol%), NaI (0.2 mmol), DABCO (2 equiv), CO (5 atm), MeCN (15 mL), 90 °C, 8 h.
b Yield of isolated product.

A proposed mechanism for the aminocarbonylation reaction is shown in Scheme [3]. The aryl iodide, in the presence of palladium chloride and the iodide ligand, undergoes oxidative addition to form intermediate II. The CO insertion step is followed by reductive elimination to regenerate the active catalyst. The reaction then proceeds via demethoxylation of the N-methoxyamide intermediate IV. The base (DABCO) plays a crucial role in releasing free methoxylamine and in the demethoxylation of the intermediate IV.
In summary, an improved palladium-catalyzed, phosphine-free carbonylation method for primary amide synthesis has been established. Methoxylamine hydrochloride undergoes sequential carbonylation and demethoxylation, and acts as an ammonia equivalent. The reaction avoids the use of expensive and moisture-/air-sensitive phosphine ligands and requires lower temperatures and shorter reaction times compared to previous protocols. This procedure offers an attractive synthetic strategy for the practical construction of aromatic primary amides.
Acknowledgment
S.T.G. is grateful to the Council of Scientific and Industrial Research (CSIR) for providing a Senior Research Fellowship.
-
References
- 1 Roy S, Roy S, Gribble GW. Tetrahedron 2012; 68: 9867
- 2a Soai K, Ookawa A, Hayashi H. J. Chem. Soc., Chem. Commun. 1983; 668
- 2b Prasad A, Kanth J, Periasamy M. Tetrahedron 1992; 48: 4623
- 3a Bose D, Jayalakshmi B. J. Org. Chem. 1999; 64: 1713
- 3b Chaudhari K, Mahajan U, Bhalerao D, Akamanchi K. Synlett 2007; 2815
- 4 Plummer BF, Menendez M, Songster M. J. Org. Chem. 1989; 54: 718
- 5a Owston NA, Parker AJ, Williams JM. J. Org. Lett. 2007; 9: 73
- 5b Foot JS, Kanno H, Giblin GM. P, Taylor RJ. K. Synlett 2002; 1293
- 6 Morera E, Ortar G. Tetrahedron Lett. 1998; 39: 2835
- 7a Schnyder A, Beller M, Mehltretter G, Nsenda T, Studer M, Indolese AF. J. Org. Chem. 2001; 66: 4311
- 7b Schnyder A, Indolese AF. J. Org. Chem. 2002; 67: 594
- 7c Wan Y, Alterman M, Larhed M, Hallberg A. J. Comb. Chem. 2003; 5: 82
- 8a Wu X.-F, Neumann H, Beller M. Chem. Asian J. 2010; 5: 2168
- 8b Wu X.-F, Schranck J, Neumann H, Beller M. ChemCatChem 2012; 4: 69
- 9 Ueda K, Sato Y, Mori M. J. Am. Chem. Soc. 2000; 122: 10722
- 10a Nielsen DU, Taaning RH, Lindhardt AT, Gøgsig TM, Skrydstrup T. Org. Lett. 2011; 13: 4454
- 10b Wu X, Wannberg J, Larhed M. Tetrahedron 2006; 62: 4665
- 10c Xu T, Alper H. Tetrahedron Lett. 2013; 54: 5496
- 11a Yanase T, Monguchi Y, Sajiki H. RSC Adv. 2012; 2: 590
- 11b Kitamura Y, Sako S, Udzu T, Tsutsui A, Maegawa T, Monguchi Y, Sajiki H. Chem. Commun. 2007; 5069
- 11c Gupte SP, Chaudhari RV. J. Catal. 1988; 114: 246
- 12a Gadge ST, Bhanage BM. J. Org. Chem. 2013; 78: 6793
- 12b Gadge ST, Khedkar MV, Lanke SR, Bhanage BM. Adv. Synth. Catal. 2012; 354: 2049
- 12c Gadge ST, Bhanage BM. Synlett 2013; 24: 981
- 13a Maitlis PM, Haynes A, James BR, Catellanic M, Chiusoli GP. Dalton Trans. 2004; 3409
- 13b Perry RJ, Wilson BD. J. Org. Chem. 1996; 61: 7482
- 14a Graham SL, Scholz TH. Tetrahedron Lett. 1990; 31: 6269
- 14b Labeeuw O, Phansavath P, Genêt J.-P. Tetrahedron Lett. 2004; 45: 7107
- 14c Keck GE, McHardy SF, Murry JA. Tetrahedron Lett. 1993; 34: 6215
- 15 Aminocarbonylation of Aryl and Heteroaryl Iodides; General Procedure To an autoclave (100 mL capacity), were added an aryl halide (1 mmol), methoxylamine hydrochloride (1.2 equiv), DABCO (2 equiv), PdCl2 (10 mol%), NaI (0.2 mmol) and MeCN (15 mL), under an inert atm. The autoclave was flushed three times with CO and then pressurized to 5 atm of CO. The mixture was stirred with a mechanical stirrer (550 rpm) at 90 °C for 8 h. The reactor was cooled to r.t., degassed carefully, opened and the reaction mixture removed. The reactor vessel was washed with EtOAc (2 × 5 mL) to remove residual product. The mixture was filtered and the filtrate washed with brine (2 × 4 mL), dried over Na2SO4, filtered and the solvent evaporated under vacuum. Purification of the residue was carried out by column chromatography (silica gel, 100–200 mesh, PE–EtOAc) to afford the corresponding product in good to excellent yield.
-
References
- 1 Roy S, Roy S, Gribble GW. Tetrahedron 2012; 68: 9867
- 2a Soai K, Ookawa A, Hayashi H. J. Chem. Soc., Chem. Commun. 1983; 668
- 2b Prasad A, Kanth J, Periasamy M. Tetrahedron 1992; 48: 4623
- 3a Bose D, Jayalakshmi B. J. Org. Chem. 1999; 64: 1713
- 3b Chaudhari K, Mahajan U, Bhalerao D, Akamanchi K. Synlett 2007; 2815
- 4 Plummer BF, Menendez M, Songster M. J. Org. Chem. 1989; 54: 718
- 5a Owston NA, Parker AJ, Williams JM. J. Org. Lett. 2007; 9: 73
- 5b Foot JS, Kanno H, Giblin GM. P, Taylor RJ. K. Synlett 2002; 1293
- 6 Morera E, Ortar G. Tetrahedron Lett. 1998; 39: 2835
- 7a Schnyder A, Beller M, Mehltretter G, Nsenda T, Studer M, Indolese AF. J. Org. Chem. 2001; 66: 4311
- 7b Schnyder A, Indolese AF. J. Org. Chem. 2002; 67: 594
- 7c Wan Y, Alterman M, Larhed M, Hallberg A. J. Comb. Chem. 2003; 5: 82
- 8a Wu X.-F, Neumann H, Beller M. Chem. Asian J. 2010; 5: 2168
- 8b Wu X.-F, Schranck J, Neumann H, Beller M. ChemCatChem 2012; 4: 69
- 9 Ueda K, Sato Y, Mori M. J. Am. Chem. Soc. 2000; 122: 10722
- 10a Nielsen DU, Taaning RH, Lindhardt AT, Gøgsig TM, Skrydstrup T. Org. Lett. 2011; 13: 4454
- 10b Wu X, Wannberg J, Larhed M. Tetrahedron 2006; 62: 4665
- 10c Xu T, Alper H. Tetrahedron Lett. 2013; 54: 5496
- 11a Yanase T, Monguchi Y, Sajiki H. RSC Adv. 2012; 2: 590
- 11b Kitamura Y, Sako S, Udzu T, Tsutsui A, Maegawa T, Monguchi Y, Sajiki H. Chem. Commun. 2007; 5069
- 11c Gupte SP, Chaudhari RV. J. Catal. 1988; 114: 246
- 12a Gadge ST, Bhanage BM. J. Org. Chem. 2013; 78: 6793
- 12b Gadge ST, Khedkar MV, Lanke SR, Bhanage BM. Adv. Synth. Catal. 2012; 354: 2049
- 12c Gadge ST, Bhanage BM. Synlett 2013; 24: 981
- 13a Maitlis PM, Haynes A, James BR, Catellanic M, Chiusoli GP. Dalton Trans. 2004; 3409
- 13b Perry RJ, Wilson BD. J. Org. Chem. 1996; 61: 7482
- 14a Graham SL, Scholz TH. Tetrahedron Lett. 1990; 31: 6269
- 14b Labeeuw O, Phansavath P, Genêt J.-P. Tetrahedron Lett. 2004; 45: 7107
- 14c Keck GE, McHardy SF, Murry JA. Tetrahedron Lett. 1993; 34: 6215
- 15 Aminocarbonylation of Aryl and Heteroaryl Iodides; General Procedure To an autoclave (100 mL capacity), were added an aryl halide (1 mmol), methoxylamine hydrochloride (1.2 equiv), DABCO (2 equiv), PdCl2 (10 mol%), NaI (0.2 mmol) and MeCN (15 mL), under an inert atm. The autoclave was flushed three times with CO and then pressurized to 5 atm of CO. The mixture was stirred with a mechanical stirrer (550 rpm) at 90 °C for 8 h. The reactor was cooled to r.t., degassed carefully, opened and the reaction mixture removed. The reactor vessel was washed with EtOAc (2 × 5 mL) to remove residual product. The mixture was filtered and the filtrate washed with brine (2 × 4 mL), dried over Na2SO4, filtered and the solvent evaporated under vacuum. Purification of the residue was carried out by column chromatography (silica gel, 100–200 mesh, PE–EtOAc) to afford the corresponding product in good to excellent yield.