

Subscribe to RSS
DOI: 10.1055/a-2759-6397
Synthesis of Tetrahydroisoquinolinoquinolines via Sequential Pd-Catalyzed N-Arylation and Pd-Catalyzed Alkene Carboamination Reactions
Authors
Funding Information The authors thank the NIH-NIGMS (GM124030) for financial support of this work.

Abstract
The synthesis of substituted tetrahydroisoquinolinoquinolines via a two-step route involving a Pd-catalyzed N-arylation reaction followed by a Pd-catalyzed alkene carboamination reaction is described. The combined transformations lead to the generation of two rings and three bonds (two C–N and one C–C) and the heterocyclic products are obtained in moderate to good yields.
Keywords
Tetrahydroisoquinolinoquinolines - Alkene carboamination - Palladium catalysis - N-arylation - Nitrogen heterocyclesIntroduction
Tetrahydroquinolines (THQs, 1) and tetrahydroisoquinolines (THIQs, 2) are important classes of bicyclic nitrogen heterocycles that have been found to possess a broad range of biological activities.[1] Tetrahydroisoquinolinoquinolines (THIQQs, 3) are a rare class of tetracyclic nitrogen heterocycles that feature a THQ core that is fused to a THIQ core ([Fig. 1]). Despite the medicinal significance of THQs and THIQs, the bioactivity of THIQQs remains largely unexplored outside of a few derivatives that contain carbonyl groups or imine groups on one or both saturated rings.[2]

Only two approaches to the synthesis of THIQQs lacking carbonyl functionality have been previously reported ([Scheme 1]). Pearson has described one example of the synthesis of the parent THIQQ ring system 3a in 67% yield via the Schmidt reaction of azide 5 that was prepared via a Mitsunobu reaction of benzyl-alcohol-substituted indanol 4 (Eq. (1)).[3] However, this transformation involves the generation and handling of a potentially hazardous alkyl azide intermediate. Schneider has described the use of asymmetric intramolecular Diels–Alder reactions of N-hex-5-enylanilines bearing ortho-hydroxyalkyl groups (e.g., 6).[4] These reactions generated products such as 3i in good yield and enantioselectivity (Eq. (2)). However, most products generated contained aryl groups at the benzylic position of the THQ ring. Thus, it is possible that the lack of available synthetic tools to prepare these compounds has limited the exploration of their properties.

We have previously described the synthesis of fused-ring tetracyclic nitrogen heterocycles 8 and 11 using fully intramolecular Pd-catalyzed alkene carboamination reactions of substrates such as 7 and 10 ([Scheme 2]).[5] The substrates 7 were prepared in moderate yield using a two-step reductive amination sequence,[5a] whereas substrates 10 were generated in situ via Pd-catalyzed N-arylation of 2-allylanilines (e.g., 9) with o-bromochlorobenzene.[5b] We reasoned that a similar strategy of employing a fully intramolecular alkene carboamination reaction could be used to access substituted THIQQs.[6] As shown in [Scheme 1], the Pd-catalyzed N-arylation of a 1-bromo-2-(but-3-en-1-yl)benzene derivative (e.g., 12a) with a 2-chlorobenzylamine derivative such as 13a would produce intermediate aniline derivative 14a, which could undergo a subsequent Pd-catalyzed intramolecular alkene carboamination reaction to afford the desired products such as 3a. This strategy uses simple starting materials that are either commercially available or can be prepared using short synthetic routes. In addition, the reaction conditions for both Pd-catalyzed N-arylation and Pd-catalyzed alkene carboamination are relatively mild and have been shown to tolerate a range of functional groups.[7] Herein, we describe our studies on the development and scope of this new two-step sequence for THIQQ synthesis.

Results and Discussion
In preliminary studies, we attempted to conduct both the Pd-catalyzed N-arylation reaction and the Pd-catalyzed alkene carboamination reaction in a one-pot process. As such, mixtures of 1-bromo-2-(but-3-en-1-yl)benzene 12a, which can be prepared in one step,[8] and 2-chlorobenzylamine 13a were treated with catalysts composed of Pd2(dba)3 and commercially available phosphine ligands that have previously shown utility for Pd-catalyzed N-arylation reactions and/or Pd-catalyzed alkene carboamination reactions ([Table 1]). Despite considerable screening, we were unable to find suitable conditions that were effective for both the N-arylation and the alkene carboamination reactions. Some ligands such as BINAP and DPE-Phos were effective for the N-arylation, but not sufficiently electron rich to facilitate oxidative addition of the less reactive aryl chloride group in 14a. Other ligands, such as S-Phos and RuPhos, produced both the N-arylated intermediate 14a and the desired product 3a. However, neither were effective for the second step and gave 3a in only modest yields, along with side products resulting from reduction of the aryl chloride. PCy3, which is an excellent ligand for the alkene carboamination step (see below), was not effective for the N-arylation, and produced mostly but-3-en-1-yl benzene via reduction of the aryl bromide.
aConditions: 1.0 equiv 12a, 1.2 equiv 13a, 1.4 equiv NaO t Bu, 1 mol% Pd2(dba)3, 2 mol% bis-phosphine or 4 mol% monodentate phosphine, toluene (0.5 M), 105 °C, 16 h.
bYields are NMR yields determined using phenanthrene as an internal standard.
cThe main side products formed in these reactions resulted from competing Heck arylation of the starting material and reduction of the chloro group in 14a.
dThe major product was but-3-en-1-yl benzene, which results from reduction of the aryl bromide.
Since we were not successful at developing the one-pot transformation, we elected to examine the synthesis of the desired tetracyclic heterocycles through a two-step sequence, where the N-arylation would be conducted first. The resulting substituted aniline derivative would then be isolated, purified, and subjected to conditions to affect the Pd-catalyzed intramolecular alkene carboamination reaction. This would allow us to separately optimize the N-arylation and alkene carboamination steps.
Prior studies by Buchwald have shown that many N-arylation reactions of primary amines are efficiently catalyzed by a mixture of Pd2(dba)3, and BINAP.[9] As such, we examined those conditions first, and found that the coupling of 12a with 13a produced the N-arylation product 14a in excellent yield (Eq. (3)).

We then sought to optimize conditions for the Pd-catalyzed alkene carboamination reaction. Our prior studies have shown that PCy3, which is handled as the air-stable HBF4 salt,[10] is an effective ligand for fully intramolecular Pd-catalyzed alkene carboaminations that form either five-membered fused-ring[5] or bridged-ring[11] products. We also found this ligand to be highly effective for the conversion of 14a to 3a, providing the desired product in 93% NMR yield ([Table 2], entry 1). Efforts to decrease reaction temperatures to 80 °C resulted in lower yields due to incomplete consumption of starting material (entry 2). DPE-Phos and Xantphos, which are good ligands for many other alkene carboamination reactions,[6] failed to produce significant amounts of product (entries 4 and 5). This is likely due to the slow oxidative addition of the aryl chloride with these ligands. The electron-rich Cy4DPE-Phos also failed to produce the desired product. In all cases examined, side products were not observed, and the reactions that did not produce significant amounts of 3a returned only unreacted starting material 14a.
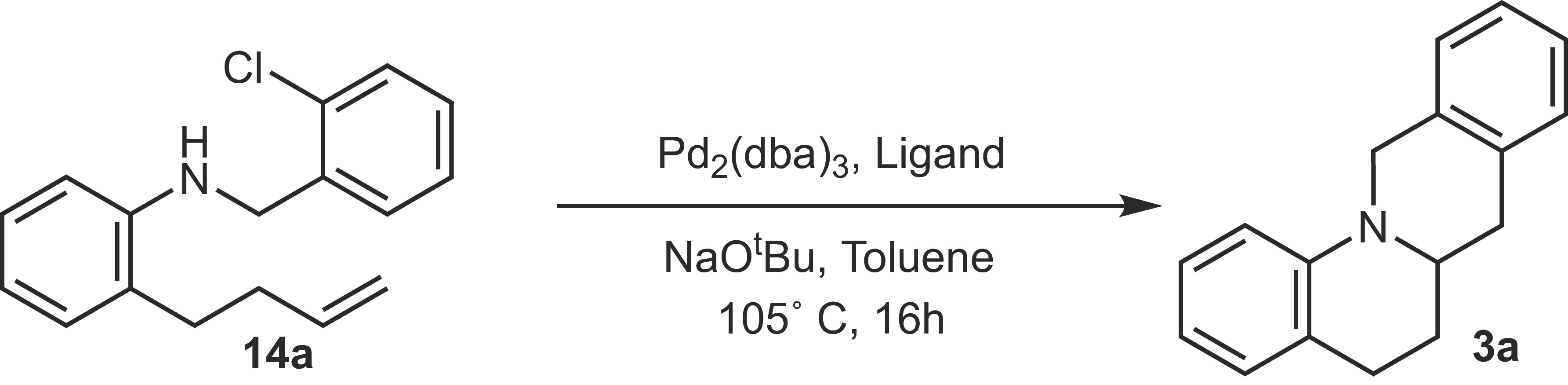 |
||
|---|---|---|
|
Entry |
Ligand |
Yield 3a (%)b |
|
1 |
PCy•HBF4 |
93 |
|
2 |
PCy•HBF4 |
59c |
|
3 |
Cy4DPE-Phos |
trace |
|
4 |
Xantphos |
trace |
|
5 |
DPE-Phos |
0% |
aConditions: 1.0 equiv 14a, 2.0 equiv NaO t Bu, 2 mol% Pd2(dba)3, 4 mol% monodentate ligand or 2 mol% bidentate ligand, Toluene (0.1 M), 105 °C, 16 h.
bYields are NMR yields determined using phenanthrene as an internal standard.
cThe reaction was conducted at 80 °C.
Having arrived at satisfactory conditions for both the N-arylation step and the alkene carboamination step, we began to explore the range of structures that could be prepared using the two-step reaction sequence. We first prepared a series of N-(2-bromobenzyl)-2-(but-3-en-1-yl)benzene derivatives 14 through Pd-catalyzed N-arylation reactions of aryl bromides 12 with 2-chlorobenzylamine derivatives 13 ([Table 3]). In general, the Pd-catalyzed N-arylation reactions proceeded smoothly, and provided the substituted N-(2-chlorobenzyl)anilines 14 in good to excellent yields. The reactions were sensitive to steric bulk near the reactive sites, as a 2-chlorobenzylamine bearing an α-methyl group was coupled with 12a to afford 14j in 60% yield. A moderate yield of 14h (66%) was also obtained in the coupling of 12a with a 2-chlorobenzylamine substituted at the ortho position. The coupling of 13a with an aryl bromide that was substituted at the allylic position also proceeded in modest yield (61%) to afford 14f.
 |
aConditions: 1.0 equiv 12, 1.2 equiv 13, 2.0 equiv NaO t Bu, 1 mol% Pd2(dba)3, 4 mol% (±)-BINAP, toluene (0.1 M), 105 °C.
bYields are isolated yields (average of two or more experiments).
We then explored the scope of the intramolecular alkene carboamination reactions using the substrates described above. As shown in [Table 4], the products of these reactions were obtained in moderate to good yields. The presence of either electron-donating groups (3e, 3h) or electron-withdrawing groups (3c, 3g) on either of the two aromatic rings was tolerated. Transformations of substrates bearing methyl groups on the internal alkene carbon (14d) or the allylic position (14f) afforded products 3d and 3f in yields that were slightly lower than those obtained with less-hindered substrates. However, the reaction of 14j, which contains a benzylic methyl group, did not proceed to completion, and produced a ca 52% yield (NMR) of the desired product 3j as a 1.3:1 mixture of diastereomers, along with ca 12% of a side product resulting from reduction of the aryl chloride, and ca 38% unreacted 14j. A small amount (32%) of 3j was isolated via preparative TLC. Efforts to employ a substrate with an internal alkene were unsuccessful; no desired product was generated.
 |
aConditions: 1.0 equiv 14, 2.0 equiv NaO t Bu, 2 mol% Pd2(dba)3, 8 mol% PCy3•HBF4, toluene (0.1 M), 105 °C.
bYields are isolated yields (average of two or more experiments).
The mechanism of the intramolecular Pd-catalyzed alkene carboamination reaction is likely analogous to that of previously reported transformations.[5] [6] As shown in [Scheme 3], the catalytic cycle is initiated by the oxidative addition of the aryl chloride 14a to Pd(0) to generate the Pd(II) complex 15a. An electron-rich ligand (PCy3) is needed to facilitate this step due to the relatively slow rate of oxidative addition of aryl chlorides relative to that of aryl bromides.[12] Complex 15a is then transformed to palladium–amido complex 16a through reaction with NaO t Bu and coordination of the alkene to the metal.[8] Intermediate 16a undergoes migratory insertion into the Pd–N bond to provide 17a, which is transformed to product 3a via reductive elimination. This reductive elimination releases the Pd(0) catalyst, which can reenter the catalytic cycle.

Conclusion
In conclusion, we have developed a two-step method (Pd-catalyzed N-arylation followed by Pd-catalyzed alkene carboamination) for the synthesis of THIQQs. The starting materials are readily synthesized or purchased, allowing for the rapid construction of fused ring nitrogen heterocycles from simple starting materials. The two-step reaction sequence generates two carbon–nitrogen bonds, one carbon–carbon bond, and two rings to produce a rare class of heterocyclic compounds whose synthesis has seldom been described.
All reactions were carried out under a nitrogen atmosphere using oven- or flame-dried glassware. All palladium complexes, phosphine ligands, and reagents were obtained from commercial sources and used without further purification unless otherwise noted. The o-bromohomoallylbenzene substrates 1-bromo-2-(but-3-en-1-yl)benzene (12a),[7] 1-bromo-2-(but-3-en-1-yl)naphthalene (12b),[13] 2-bromo-1-(but-3-en-1-yl)-4-(trifluoromethyl)benzene (12c),[14] 1-bromo-2-(3-methylbut-3-en-1-yl)benzene (12d),[15] 1-bromo-2-(but-3-en-1-yl)-4-methoxybenzene (12e),[16] 1-bromo-2-(2-methylbut-3-en-1-yl)benzene (12f),[17] 3-bromo-4-(but-3-en-1-yl)pyridine (12g),[18] and o-chlorobenzylamines (2-chloro-3,4-dimethoxyphenyl)methanamine (13c),[19] and 1-(2-chlorophenyl)ethan-1-amine (13d),[20] were prepared according to previously reported procedures. Toluene was purified using a GlassContour solvent purification system. All yields refer to isolated compounds that are estimated to be ≥95% pure as judged by 1H NMR analysis unless otherwise noted. The yields reported in the experimental section describe the results of a single experiment, whereas yields reported in [Tables 3] and [4] are average yields of two or more experiments. Thus, the yields reported in the experimental section may differ from those shown in [Tables 3] and [4].
Procedures
General Procedure 1: Pd-Catalyzed N-Arylation Reactions
A flame-dried Schlenk tube equipped with a stir bar was cooled under a stream of N2 and charged with Pd2(dba)3 (1 mol%), BINAP (4 mol%), and NaOtBu (0.6 mmol). The tube was capped with a rubber septum, then was evacuated and backfilled with N2 gas three times. The appropriate o-bromohomoallylbenzene 12 (0.3 mmol) and the appropriate o-chlorobenzylamine 13 (0.36 mmol) were dissolved in toluene (3 mL, 0.1 M) and added via syringe. The reaction mixture was heated to 105 °C for 16 h unless otherwise specified. The mixture was then cooled to room temperature, diluted with either Et2O or dichloromethane, and filtered through a packed pad of Celite. Additional Et2O or dichloromethane was eluted through the Celite to ensure minimal compound loss. The resulting solution was concentrated in vacuo, and the crude product 14 was purified by flash chromatography on silica gel.
2-(But-3-en-1-yl)-N-(2-chlorobenzyl)aniline (14a)
General procedure 1 was used for the coupling of 12a (63.3 mg, 0.3 mmol) with 13a (51.0 mg, 0.36 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (81 mg, 100%) as a pale yellow solid, mp 47–48 °C. 1H NMR (500 MHz, CDCl3) δ 7.43–7.36 (m, 2 H), 7.24–7.18 (m, 2 H), 7.12–7.05 (m, 2 H), 6.71 (t, J = 7.5 Hz, 1 H), 6.57 (d, J = 8.2 Hz, 1 H), 5.99–5.87 (m, 1 H), 5.11 (d, J = 17.9 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.49 (s, 2 H), 4.14 (s, 1 H), 2.63 (t, J = 7.6 Hz, 2 H), 2.47–2.38 (m, 2 H). 13C NMR (126 MHz, CDCl3) δ 145.1, 138.1, 136.7, 133.3, 129.6, 129.1, 129.0, 128.4, 127.3, 126.9, 125.6, 117.5, 115.1, 110.7, 46.0, 32.8, 30.7. IR (film) 3446, 3069, 2925, 1604, 1509 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C17H18ClN 272.1180; found 272.1172.
2-(But-3-en-1-yl)-N-(2-chlorobenzyl)naphthalen-1-amine (14b)
General procedure 1 was used for the coupling of 12b (78.4 mg, 0.3 mmol) with 13a (51 mg, 0.36 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (89 mg, 92%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 8.19 (d, J = 8.6 Hz, 1 H), 7.80 (d, J = 7.6 Hz, 1 H), 7.52 (d, J = 8.4 Hz, 1 H), 7.48 (t, J = 7.1 Hz, 1 H), 7.46–7.40 (m, 2 H), 7.37 (m, 1 H), 7.28 (d, J = 8.4 Hz, 1 H), 7.25–7.20 (m, 2 H), 5.91–5.81 (m, 1 H), 5.05 (d, J = 17.4 Hz, 1 H), 5.00 (d, J = 9.7 Hz, 1 H), 4.39 (s, 2 H), 3.92 (s, 1 H), 2.75 (t, J = 7.6 Hz, 2 H), 2.35–2.30 (m, 2 H). 13C NMR (151 MHz, CDCl3) δ 141.6, 138.0, 137.7, 133.8, 133.7, 130.6, 130.2, 129.7, 129.4, 128.8, 128.5, 128.2, 127.2, 125.8, 125.3, 123.6, 123.5, 115.5, 52.6, 35.2, 31.2. IR (film) 3378, 3083, 2946, 1606, 1511 cm−1; HRMS (ESI TOF) m/z: [M + H]+ calcd for C21H20ClN, 322.1364; found, 322.1362.
2-(But-3-en-1-yl)-N-(2-chlorobenzyl)-5-(trifluoromethyl)aniline (14c)
General procedure 1 was used for the coupling of 12c (83.7 mg, 0.3 mmol) with 13a (51.0 mg, 0.3 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (79.5 mg, 78%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.48–7.40 (m, 1 H), 7.40–7.33 (m, 1 H), 7.27–7.23 (m, 2 H), 7.15 (d, J = 7.7 Hz, 1 H), 6.95 (d, J = 6.0 Hz, 1 H), 6.80 (s, 1 H), 5.95–5.83 (m, 1 H), 5.06 (m, 2 H), 4.50 (d, J = 5.6 Hz, 2 H), 4.25 (s, 1 H), 2.63 (t, J = 7.8 Hz, 2 H), 2.41 (q, J = 6.4 Hz, 2 H). 13C NMR (151 MHz, CDCl3) δ 145.6, 137.7, 136.0, 133.8, 130.0, 129.8, 129.6, 129.4, 129.4, 129.1, 127.3, 125.6, 123.8, 115.8, 114.2, 114.2, 114.2, 114.2, 107.0, 106.9, 106.9, 106.9, 46.1, 32.5, 30.7. IR (film) 3364, 3115, 2918, 1517 cm−1; HRMS (ESI TOF) m/z: [M + H]+ calcd for C18H17ClF3N, 340.1080; found, 340.1052.
N-(2-Chlorobenzyl)-2-(3-methylbut-3-en-1-yl)aniline (14d)
General procedure 1 was used for the coupling of 12d (45.0 mg, 0.2 mmol) with 13a (34.0 mg, 0.24 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (48 mg, 88%) as a white solid, mp 50–51 °C. 1H NMR (500 MHz, CDCl3) δ 7.42–7.36 (m, 2 H), 7.24–7.19 (m, 2 H), 7.11–7.05 (m, 2 H), 6.71 (t, J = 7.3 Hz, 1 H), 6.56 (d, J = 8.1 Hz, 1 H), 4.76 (d, J = 7.1 Hz, 2 H), 4.50 (s, 2 H), 4.14 (s, 1 H), 2.66 (t, J = 8.7 Hz, 2 H), 2.35 (t, J = 8.4 Hz, 2 H), 1.79 (s, 3 H). 13C NMR (126 MHz, CDCl3) δ 145.6, 129.6, 129.1, 128.9, 128.4, 127.2, 126.9, 110.3, 46.0, 36.7, 29.7, 22.7. IR (film) 3321, 3106, 2862, 1601, 1488 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C18H20ClN, 285.1284; found, 285.1275.
2-(But-3-en-1-yl)-N-(2-chlorobenzyl)-4-methoxyaniline (14e)
General procedure 1 was used for the coupling of 12d (72.3 mg, 0.3 mmol) with 13a (51.0 mg, 0.3 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (67 mg, 74%) as an orange oil 1H NMR (600 MHz, CDCl3) δ 7.42–7.34 (m, 2 H), 7.24–7.18 (m, 2 H), 6.71 (d, J = 2.6 Hz, 1 H), 6.66 (dd, J = 8.7 Hz, 1 H), 6.51 (d, J = 8.5 Hz, 1 H), 5.96–5.87 (m, 1 H), 5.09 (dt, J = 17.1, 1.7 Hz, 1H), 5.01 (dq, J = 10.1, 1.0 Hz, 1H), 3.84 (s, 1 H), 3.74 (s, 3 H), 2.64–2.58 (m, 2 H), 2.44–2.38 (m, 2 H). 13C NMR (151 MHz, CDCl3) δ 152.0, 139.4, 138.0, 137.0, 133.3, 129.5, 129.1, 128.3, 127.7, 126.9, 116.0, 115.2, 112.1, 111.5, 55.7, 46.8, 32.9, 30.8. IR (film) 3437, 2929, 1640, 1508, 1468, 1224 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C18H20ClNO, 301.1233; found, 302.1221.
N-(2-Chlorobenzyl)-2-(2-methylbut-3-en-1-yl)aniline (14f)
General procedure 1 was used for the coupling of 12f (113.1 mg, 0.5 mmol) with 13a (85 mg, 0.6 mmol). The crude product was purified via column chromatography using 3% EtOAc in hexanes as the eluent to give the title compound (83 mg, 61%) as an off-white solid, mp 49–50 °C. 1H NMR (401 MHz, CDCl3) δ 7.42–7.33 (m, 2 H), 7.21 (dd, J = 5.9, 3.5 Hz, 2 H), 7.12–7.00 (m, 2 H), 6.69 (t, J = 7.4, 1 H), 6.56 (d, J = 8.1 Hz, 1 H), 5.91–5.78 (m, 1 H), 5.02–4.90 (m, 2 H), 4.48 (s, 2 H), 4.21 (s, 1 H), 2.66–2.42 (m, 3 H), 1.06 (d, J = 6.2 Hz, 3 H). 13C NMR (151 MHz, CDCl3) δ 145.4, 144.0, 136.8, 133.2, 130.7, 129.5, 129.0, 128.3, 127.3, 126.9, 124.7, 117.3, 112.9, 110.8, 46.0, 38.8, 36.9, 19.8. IR (film) 3470, 2930, 1510 cm−1; HRMS (ESI TOF) m/z: [M + H]+ calcd for C18H20ClN, 285.1284; found, 285.1279.
2-(But-3-en-1-yl)-N-(2-chloro-4-fluorobenzyl)aniline (14g)
General procedure 1 was used for the coupling of 12a (105.6 mg, 0.5 mmol) with 13b (95.8 mg, 0.6 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (114 mg, 79%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.35 (dd, J = 8.6, 6.1 Hz, 1 H), 7.16 (dd, J = 8.4, 2.6 Hz, 1 H), 7.10–7.06 (m, 2 H), 6.93 (td, J = 8.3, 2.6 Hz, 1 H), 6.75–6.69 (m, 1 H), 6.52 (dd, J = 8.6, 1.2 Hz, 1 H), 5.97–5.87 (m, 1 H), 5.07 (m, 2 H), 4.44 (d, J = 5.3 Hz, 2 H), 4.11 (s, 1 H), 2.65–2.59 (m, 2 H), 2.47–2.36 (m, 2 H). 13C NMR (176 MHz, CDCl3) δ 162.4, 160.9, 144.9, 138.1, 132.6, 129.1, 127.3, 125.7, 117.7, 117.0, 115.2, 114.1, 110.6, 53.4, 45.4, 32.8, 30.7. IR (film) 3391, 2984, 1601 cm−1; HRMS (ESI TOF) m/z: [M + H]+ calcd for C17H17ClFN, 290.1104; found, 290.1112.
2-(But-3-en-1-yl)-N-(2-chloro-3,4-dimethoxybenzyl)aniline (14h)
General procedure 1 was used for the coupling of 12a (211.1 mg, 1.0 mmol) with 13c (242 mg, 1.2 mmol). The crude product was purified via column chromatography using 10% EtOAc in hexanes as the eluent to give the title compound (214 mg, 64%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.13–7.05 (m, 3 H), 6.78 (d, J = 8.6 Hz, 1 H), 6.70 (t, J = 7.4 Hz, 1 H), 6.58 (d, J = 8.0 Hz, 1 H), 5.97–5.85 (m, 1 H), 5.10 (dd, J = 151.7, 16.9 Hz, 2 H), 4.41 (s, 2 H), 4.09 (s, 1 H), 3.89 (s, 3 H), 3.86 (s, 3 H), 2.61 (t, J = 7.0 Hz, 2 H), 2.44–2.36 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 152.8, 145.7, 145.2, 138.1, 129.5, 129.0, 127.7, 127.2, 125.6, 123.8, 117.4, 115.1, 110.7, 110.4, 60.6, 56.1, 45.8, 32.8, 30.7. IR (film): 3438, 2930, 2836, 1447, 1290, 1263 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C19H22ClNO2, for C19H22ClNO2, 331.1339; found, 331.1335.
N-Benzyl-4-(but-3-en-1-yl)pyridin-3-amine (14i)
General procedure 1 was used for the coupling of 12g (424.1 mg, 2.0 mmol) with 13a (339 mg, 2.4 mmol). The crude product was purified via column chromatography using 10% EtOAc, 2% triethylamine in hexanes as the eluent to give the title compound (525 mg, 97% yield) as a pink solid, mp 74–76 °C. 1H NMR (500 MHz, CDCl3) δ 7.98–7.91 (m, 2H), 7.42–7.32 (m, 2H), 7.28–7.16 (m, 2H), 6.95 (d, J = 4.8 Hz, 1H), 5.92–5.80 (m, 1H), 5.02 (dd, J = 10.0, 1.8 Hz, 2H), 4.50 (d, J = 5.9 Hz, 2H), 4.14 (s, 1H), 2.58 (dd, J = 9.0, 6.5 Hz, 2H), 2.44–2.36 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 141.3, 139.6, 137.1, 135.9, 133.6, 133.4, 133.1, 129.8, 129.2, 128.8, 127.1, 123.3, 115.8, 45.9, 31.7, 29.6. IR (film) 3275, 3075, 1566 cm−1; HRMS (APCI+) m/z: [M + H]+ calcd for C16H17ClN2, 273.1153; found 273.1153.
2-(But-3-en-1-yl)-N-(1-(2-chlorophenyl)ethyl)aniline (14j)
General procedure 1 was used for the coupling of 12a (105 mg, 0.5 mmol) with 13d (93 mg, 0.6 mmol). The crude product was purified via column chromatography using 5% EtOAc and 1% triethylamine in hexanes as the eluent to give the title compound (90 mg, 66%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.40 (ddd, J = 18.9, 7.1, 2.3 Hz, 2 H), 7.18 (tt, J = 7.4, 5.4 Hz, 2 H), 6.96 (td, J = 7.7, 1.6 Hz, 1 H), 6.65 (t, J = 7.4 Hz, 1 H), 6.23 (d, J = 8.1 Hz, 1 H), 6.05–5.93 (m, 1 H), 5.20–5.13 (m, 1 H), 5.08 (dd, J = 10.3, 1.8 Hz, 1 H), 4.94 (q, J = 6.6 Hz, 1 H), 4.03 (s, 1 H), 2.69 (t, J = 7.8 Hz, 2 H), 2.57–2.42 (m, 2 H), 1.56 (d, J = 6.6 Hz, 4 H).13C NMR (126 MHz, cdcl3) δ 144.1, 142.1, 138.3, 132.5, 129.7, 128.8, 128.0, 127.4, 127.2, 126.6, 125.1, 117.2, 115.2, 111.4, 50.3, 32.8, 30.8, 23.2. IR (film) 2926, 1605, 1508; HRMS (ESI TOF) m/z: [M + H]+ calcd for C18H20ClN, C18H20ClN, 285.1284; found, 285.1277.
N-(2-Chlorobenzyl)-2-(pent-3-en-1-yl)aniline (14k)
General procedure 1 was used for the coupling of 12h (113.1 mg, 0.5 mmol) 13a (85 mg, 0.6 mmol). The crude product was purified via column chromatography using 3% EtOAc in hexanes to give the title compound (110 mg, 80%) as a colorless oil. This material was judged to be a ca 2.3:1 mixture of Z:E isomers. 1H NMR (600 MHz, CDCl3) δ 7.42–7.35 (m, 3 H), 7.24–7.19 (m, 3 H), 7.11–7.04 (m, 3 H), 6.74–6.67 (m, 2 H), 6.58–6.52 (m, 2 H), 5.54–5.50 (m, 1 H), 5.52–5.46 (m, 2 H), 4.48 (d, J = 5.2 Hz, 3 H), 4.15 (s, 2 H), 2.57 (dd, J = 9.1, 6.4 Hz, 3 H), 2.45–2.38 (m, 2 H), 2.37–2.31 (m, 1 H), 1.66 (d, J = 4.9 Hz, 1 H), 1.57 (d, J = 4.7 Hz, 3 H). 13C NMR (176 MHz, CDCl3) δ 145.2, 145.2, 136.8, 136.8, 133.3, 130.7, 129.6, 129.6, 129.6, 129.1, 129.1, 129.0, 128.4, 128.4, 127.2, 127.2, 127.0, 126.0, 125.9, 125.7, 124.8, 117.5, 117.5, 110.6, 110.6, 46.0, 45.9, 31.8, 31.4, 31.1, 26.0, 18.0, 12.8. HRMS (ESI TOF) m/z: [M]+ calcd for C18H20ClN, 285.1284; found, 285.1286.
General Procedure 2: Pd-Catalyzed Alkene Carboamination Reactions
A flame-dried Schlenk tube equipped with a stir bar was cooled under a stream of N2 and charged with the Pd2(dba)3 (2 mol%), PCy3HBF4 (8 mol%), and NaOtBu (0.6 mmol). The tube was capped with a rubber septum, then was evacuated and backfilled with N2 gas three times. The appropriate substrate 14 (0.3 mmol) was dissolved in toluene (3 mL, 0.1 M) and added via syringe. The reaction mixture was heated to 105 °C for 16 h unless otherwise specified. The mixture was then cooled to room temperature, diluted with either Et2O or dichloromethane and filtered through a packed pad of Celite. Additional Et2O or dichloromethane was eluted through the Celite to ensure minimal compound loss. The resulting solution was concentrated in vacuo, and the crude product was purified by flash chromatography on silica gel.
6,6a,7,12-Tetrahydro-5H-isoquinolino[2,3-a]quinoline (3a)
General procedure 2 was used for the cyclization of 14a (81.5 mg, 0.3 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (59.5 mg, 85%) as a pale yellow oil 1H NMR (500 MHz, CDCl3) δ 7.24–7.12 (m, 5 H), 7.02 (d, 1 H), 6.87 (d, J = 8.2 Hz, 1 H), 6.71 (t, 1 H), 4.79 (d, J = 15.6 Hz, 1 H), 4.24 (d, J = 15.6 Hz, 1 H), 3.37 (s, 1 H), 3.00–2.77 (m, 3 H), 2.76–2.68 (m, 1 H), 2.15–2.05 (m, 1 H), 2.00–1.89 (m, 1 H). 13C NMR (126 MHz, CDCl3) δ 146.1, 134.8, 134.1, 128.4, 128.3, 127.2, 126.4, 126.2, 126.0, 125.3, 117.3, 112.2, 52.9, 50.1, 37.4, 29.6, 26.7. IR (film) 3023, 2929, 2838, 2360, 1602, 1495 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C17H17N, 235.1361; found, 235.1358.
8,8a,9,14-Tetrahydro-7H-benzo[h]isoquinolino[2,3-a]quinoline (3b)
General procedure 2 was used for the cyclization of 14b (81.2 mg, 0.25 mmol, 1 equiv). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (0.39 mg, 54% yield) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ 8.06–8.02 (m, 1 H), 7.81–7.75 (m, 1 H), 7.44–7.36 (m, 3 H), 7.28–7.17 (m, 4 H), 7.07 (d, J = 7.4 Hz, 1 H), 4.52 (s, 1 H), 4.24 (d, J = 15.7 Hz, 1 H), 3.53–3.46 (m, 1 H), 3.30 (d, J = 16.2 Hz, 1 H), 3.12 (m, 1 H), 2.96 (dd, J = 16.2 Hz, 1 H), 2.88 (d, J = 16.6 Hz, 1 H), 2.08–1.94 (m, 2 H). 13C NMR (151 MHz, CDCl3) δ 143.9, 135.0, 134.5, 133.7, 128.9, 128.3, 127.9, 126.5, 126.0, 125.7, 125.2, 124.9, 123.8, 121.8, 56.7 (br), 55.1 (br), 35.8, 28.5, 25.6 (br). Note: some aliphatic carbon signals were broadened due to slow conformational interconversion resulting from steric interactions of the naphthyl ring. IR (film) 3046.79, 2931.81, 2823.20, 2790.64 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C21H19N, 285.1517; found, 285.1520.
2-(Trifluoromethyl)-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3c)
General procedure 2 was used for the cyclization of 14c (92.6 mg, 0.27 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (0.725 mg, 88%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.25–7.17 (m, 3 H), 7.16 (d, J = 7.4 Hz, 1 H), 7.09 (d, J = 7.6 Hz, 1 H), 7.00 (s, 1 H), 6.94 (d, J = 7.7 Hz, 1 H), 4.75 (d, J = 15.5 Hz, 1 H), 4.31 (d, J = 15.4 Hz, 1 H), 3.49–3.39 (m, 1 H), 2.99–2.92 (m, 1 H), 2.92–2.84 (m, 1 H), 2.84–2.79 (m, 1 H), 2.80–2.72 (m, 1 H), 2.15–2.06 (m, 1 H), 2.00–1.88 (m, 1 H). 13C NMR (151 MHz, CDCl3) δ 146.3, 134.8, 133.7, 129.6, 128.9, 128.5, 128.5, 126.6, 126.6, 126.5, 125.8, 124.0, 113.7, 113.6, 113.6, 113.6, 108.1, 108.1, 108.1, 108.1, 52.9, 49.9, 37.5, 29.9, 29.2, 26.7. IR (film) 2926, 2858, 1326, 1118; HRMS (ESI TOF) m/z: [M]+ calcd for C18H17F3N, C18H17F3N, 303.1235; found, 303.1242.
6a-Methyl-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3d)
General procedure 2 was used for the cyclization of 14d (111 mg, 0.39 mmol, 1 equiv). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (52.3 mg, 54%) as a red-orange oil.1H NMR (500 MHz, CDCl3) 1H NMR (700 MHz, CDCl3) δ 7.27–7.13 (m, 5 H), 7.07 (d, J = 7.3 Hz, 1 H), 6.83 (d, J = 8.2 Hz, 1 H), 6.72 (t, J = 7.4 Hz, 1 H), 4.70 (d, J = 15.9 Hz, 1 H), 4.18 (d, J = 15.9 Hz, 1 H), 3.07 (d, J = 15.4 Hz, 1 H), 2.97–2.90 (m, 1 H), 2.79–2.73 (m, 1 H), 2.67 (d, J = 15.4 Hz, 1 H), 2.04–1.97 (m, 1 H), 1.93–1.87 (m, 1 H), 1.05 (s, 3 H). 13C NMR (176 MHz, CDCl3) δ 145.3, 133.60, 133.59, 128.8, 128.6, 127.2, 126.2, 126.1, 125.9, 124.2, 116.8, 112.0, 51.8, 46.8, 43.6, 36.9, 24.6, 20.4. IR (film) 3027, 2926, 1601, 1494 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C18H20N, 249.1517; found, 249.1508
3-Methoxy-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3e)
General procedure 2 was used for the cyclization of 14e (90.5 mg, 0.3 mmol). The crude product was purified via column chromatography using 3% EtOAc in hexanes as the eluent to give the title compound (43.0 mg, 81%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) 1H NMR (700 MHz, CDCl3) δ 7.18 (d, J = 3.7 Hz, 3 H), 7.13 (d, J = 7.4 Hz, 1 H), 6.83 (d, J = 8.9 Hz, 1 H), 6.77–6.72 (m, 1 H), 6.65 (s, 1 H), 4.76 (d, J = 15.5 Hz, 1 H), 4.15 (d, J = 15.5 Hz, 1 H), 3.77 (s, 3 H), 3.25 (s, 1 H), 2.96–2.83 (m, 2 H), 2.80 (d, J = 15.8 Hz, 1 H), 2.70 (d, J = 15.6 Hz, 1 H), 2.08 (dd, J = 13.1, 5.2 Hz, 1 H), 1.96–1.88 (m, 1 H). IR (film) 2929.20, 2849.39, 1500.44, 1243.04; HRMS (ESI TOF) m/z: [M + H]+ calcd for C18H20NO, 265.1467; found, 265.1466.
6-Methyl-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3f)
General procedure 2 was used for the cyclization of 14f (48.0 mg, 0.168 mmol). The crude product was purified via column chromatography using 35% dichloromethane in hexanes to give the title compound (27.3 mg, 65%) as a red-orange oil. This material was determined to be a ca 1.3:1 mixture of diastereomers as judged by 1H NMR analysis. Data are for the mixture. 1H NMR (500 MHz, CDCl3) 1H NMR (600 MHz, CDCl3) δ 7.23–7.14 (m, 7 H), 7.14–7.08 (m, 1 H), 7.01 (ddd, J = 12.8, 7.3, 1.6 Hz, 1H), 6.86 (d, J = 8.2 Hz, 1 H), 6.79 (d, J = 8.2 Hz, 0.75 H), 6.68 (m, 1H), 4.84 (d, J = 7.9 Hz, 0.81 H), 4.81 (d, J = 8.2 Hz, 1 H), 4.36 (d, J = 16.0 Hz, 0.79 H), 4.26 (d, J = 15.7 Hz, 1 H), 3.54 (dt, J = 11.8, 3.8 Hz, 0.74 H), 3.04–2.98 (m, 2 H), 2.91 (dd, J = 15.6, 11.8 Hz, 1 H), 2.87–2.68 (m, 4 H), 2.64–2.55 (m, 2 H), 2.32–2.24 (m, 0.76 H), 2.04–1.95 (m, 1 H), 1.13 (d, J = 6.9 Hz, 2.27 H), 1.11 (d, J = 6.7 Hz, 3 H). 13C NMR (151 MHz, CDCl3) δ 145.7, 145.0, 135.3, 134.8, 134.5, 134.1, 128.9, 128.9, 128.7, 128.4, 127.2, 127.1, 126.3, 126.2, 126.2, 126.1, 126.0, 125.9, 124.0, 123.6, 117.2, 116.7, 112.3, 111.6, 59.8, 56.8, 50.6, 50.1, 35.7, 35.2, 33.7, 33.1, 31.0, 30.4, 19.3, 15.9. IR (film) 3027, 2925, 1603, 1494 cm−1; HRMS (ESI TOF) m/z: [M + H]+ calcd for C18H19N, 250.1596; found, 250.1569
9-Fluoro-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3g)
General procedure 2 was used for the cyclization of 14g (93.0 mg, 0.32 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (54.0 mg, 66%) as a dark red oil. 1H NMR (600 MHz, CDCl3) δ 7.19–7.11 (m, 2 H), 7.03 (d, J = 5.8 Hz, 1 H), 6.94–6.87 (m, 1 H), 6.84 (d, J = 8.5 Hz, 2 H), 6.72 (t, J = 6.8 Hz, 1 H), 4.75 (d, J = 15.4 Hz, 1 H), 4.19 (d, J = 15.4 Hz, 1 H), 3.38–3.31 (m, 1 H), 2.97–2.90 (m, 1 H), 2.90–2.82 (m, 1 H), 2.80–2.68 (m, 2 H), 2.09 (dd, J = 13.1, 6.6 Hz, 1 H), 1.97–1.88 (m, 1 H). 13C NMR (176 MHz, CDCl3) δ 161.9, 160.5, 145.9, 136.8, 136.8, 129.7, 128.4, 127.8, 127.8, 127.2, 125.4, 117.5, 114.7, 114.6, 113.3, 113.2, 112.3, 52.7, 49.7, 37.3, 29.4, 26.5. IR (film) 3022, 2926, 2847, 1602, 1276 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C17H17FN, 253.1267; found, 254.1267.
8,9-Dimethoxy-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3h)
General procedure 2 was used for the cyclization of 14h (60.0mg, 0.18 mmol). The crude product was purified via column chromatography using 5% EtOAc in hexanes as the eluent to give the title compound (35.5 mg, 63%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.17–7.12 (m, 1 H), 7.02 (d, J = 6.3 Hz, 1 H), 6.90 (d, J = 8.4 Hz, 1 H), 6.83 (dd, J = 15.6, 8.3 Hz, 2 H), 6.70 (t, J = 7.3 Hz, 1 H), 4.73 (d, J = 15.2 Hz, 1 H), 4.17 (d, J = 15.1 Hz, 1 H), 3.87 (s, 3 H), 3.83 (s, 3 H), 3.31–3.24 (m, 1 H), 3.04 (dd, J = 16.2, 3.1 Hz, 1 H), 2.90–2.82 (m, 1H), 2.76–2.64 (m, 2 H), 2.15–2.07 (m, 1 H), 2.01–1.92 (m, 1 H). 13C NMR (151 MHz, CDCl3) δ 150.6, 146.2, 146.1, 129.2, 128.4, 127.4, 127.1, 125.5, 121.7, 117.3, 112.4, 110.6, 77.2, 77.0, 76.8, 60.3, 55.9, 52.75, 49.8, 31.6, 29.6, 26.6. IR (film) 2934, 2834, 1603, 1497, 1280 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C19H21NO2, 295.1572; found 295.1566.
6,6a,7,12-Tetrahydro-5H-isoquinolino[2,3-a][1,7]naphthyridine (3i)
General procedure 2 was used for the cyclization of (14i) (100 mg, 0.366 mmol). The crude product was purified via column chromatography using 1% EtOAc in hexanes as the eluent to give the title compound (40 mg, 46%) as a dark brown oil. 1H NMR (600 MHz, CDCl3) δ 8.17 (s, 1H), 7.93 (d, J = 4.7 Hz, 1H), 7.27–7.12 (m, 4H), 6.91 (d, J = 4.6 Hz, 1H), 4.83 (d, J = 15.5 Hz, 1H), 4.29 (d, J = 15.5 Hz, 1H), 3.45–3.37 (m, 1H), 2.92 (dd, J = 15.6, 11.1 Hz, 1H), 2.88–2.80 (m, 2H), 2.72 (dt, J = 16.2, 5.1 Hz, 1H), 2.12 (dq, J = 14.7, 4.9 Hz, 1H).13C NMR (151 MHz, c CDCl3) δ 142.6, 138.6, 134.3, 133.8, 133.3, 132.6, 128.3, 126.4, 126.4, 126.3, 122.8, 52.5, 49.5, 36.9, 28.6, 25.9. IR (film) 1320, 1420, 1500 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C16H16N2, 236.1313; found 236.1319.
12-Methyl-6,6a,7,12-tetrahydro-5H-isoquinolino[2,3-a]quinoline (3j)
General procedure 2 was used for the cyclization of 14j (50 mg, 0.124 mmol). This reaction did not proceed to completion, and afforded a mixture of unreacted starting material (ca 38%), a major product diastereomer (ca 38%), a minor product diastereomer (ca 12%) and a side product resulting from reduction of the aryl chloride in the starting material (ca 12%). This mixture proved to be difficult to separate, and after attempted purification via column chromatography using 1% EtOAc in hexanes as the eluent led to only modest improvement in product purity. In order to obtain a pure sample of material for characterization, the mixture was purified by preparatory TLC to afford 10 mg (32%) of the major diastereomer of the title compound. 1H NMR (500 MHz, CDCl3) δ 7.25–7.05 (m, 5 H), 6.98 (dd, J = 7.2, 1.6 Hz, 1 H), 6.86 (d, J = 8.3 Hz, 1 H), 6.62 (td, J = 7.3, 1.1 Hz, 1 H), 5.14 (q, J = 6.7 Hz, 1 H), 3.70 (dq, J = 7.9, 2.8 Hz, 1 H), 2.94 (dd, J = 15.9, 11.3 Hz, 1 H), 2.82–2.68 (m, 3 H), 2.11–2.04 (m, 1 H), 1.95 (ddd, J = 8.4, 6.1, 4.4 Hz, 1 H), 1.47 (d, J = 6.6 Hz, 3 H). 13C NMR (126 MHz, CDCl3) δ 144.7, 139.6, 134.3, 128.8, 128.6, 127.2, 126.9, 126.0, 125.8, 125.2, 116.2, 112.4, 52.8, 47.3, 36.9, 28.8, 26.2, 18.8. IR (film) 3064, 1639, 1494 cm−1; HRMS (ESI TOF) m/z: [M]+ calcd for C16H16N, 249.1517; found 249.1510.
Contributors’ Statement
Kathryn G. Berges: Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review & editing. Gabriel A. Gonzalez: Conceptualization, Formal analysis, Investigation, Methodology, Supervision, Writing - review & editing. Glorimar Miranda-Mendez: Investigation, Methodology, Writing - review & editing. John P. Wolfe: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing - original draft, Writing - review & editing.
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgment
The authors congratulate Dani Schultz as a 2025 recipient of the Dr. Margaret Faul Women In Chemistry Award!
-
References
- 1a Yatav P, Kumar A, Althagafi I, Nemaysh V, Rai R, Pratap R. Curr Top Med Chem 2021; 21: 1587-1622
- 1b Faheem, Kumar BK, KVGC S, Chander S, Kunjiappan S, Murugesan S. RSC Adv 2021; 11: 12254-12287
- 1c Khadem S, Marles RJ. Nat Prod Res 2025; 399: 182-194
- 2a Ishihara Y, Kiyota Y, Goto G. Chem Pharm Bull 1990; 38: 3024-3030
- 2b Nishijima K, Shinkawa T, Ito M. et al. Eur J Med Chem 1998; 33: 763-774
- 3 Pearson WH, Fang WK. J Org Chem 2000; 65: 7158-7174
- 4a Hofmann F, Gartner C, Kretzschmar M, Schneider C. Synthesis 2022; 54: 1055-1080
- 4b Kretzschmar M, Hofmann F, Moock D, Schneider C. Angew Chem Int Ed 2018; 57: 4774-4778
- 5a Alicea J, Wolfe JP. J Org Chem 2014; 79: 4212-4217
- 5b Culberson MR, Dong S, Wolfe JP. Org Lett 2025; 27: 6445-6448
- 6 This work derives from and extends from the work described in Dr. Gabriel Gonzalez’s Ph.D. Dissertation: Gonzalez, Gabrial A., Ph.D. Dissertation, University of Michigan, 2024. https://www.proquest.com/dissertations-theses/studies-on-development-palladium-catalyzed-alkene/docview/3065166871/se-2
- 7 Wolfe JP. Top Heterocycl Chem 2013; 1-38
- 8 Wolfe JP, Rennels RA, Buchwald SL. Tetrahedron 1996; 52: 7525-7546
- 9a Wolfe JP, Wagaw S, Buchwald SL. J Am Chem Soc 1996; 118: 7215-7216
- 9b Wolfe JP, Buchwald SL. J Org Chem 2000; 65: 1144-1157
- 9c Schulummer B, Scholz U. Adv Synth Catal 2004; 346: 1599-1626
- 9d Muci AR, Buchwald SL. Top Curr Chem 2002; 219: 131-209
- 10 Netherton MR, Fu GC. Org Lett 2001; 3: 4295-4298
- 11 Schultz DM, Wolfe JP. Org Lett 2011; 13: 2962-2965
- 12a Grushin VV, Alper H. Chem Rev 1994; 94: 1047-1062
- 12b Portnoy M, Milstein D. Organometallics 1993; 12: 1665-1673
- 13 Tietze LF, Dufert MA, Hungerland T, Oum K, Lenzer T. Chem Eur J 2011; 17: 8452-8461
- 14 Cho SH, Hartwig JF. J Am Chem Soc 2013; 135: 8157-8160
- 15 Athelstan J, Beckwit L, Sendaba G. Aust J Chem 1992; 45: 289-308
- 16 Taber DF, Malcollm SC. J Org Chem 2001; 66: 944-953
- 17 Minatti A, Buchwald SL. Org Lett 2008; 10: 2721-2724
- 18 Zhang Z, Dwoskin LP, Crooks PA. Tetrahedron Lett 2011; 52: 2667-2669
- 19 Beyer TA, Chambers RJ, Lam K, Li M, Morrell AI, Thompson DD. WO Patent WO2005061497A1 2005
- 20 Xie Y, Zhang Q, Zhang H, Luo G, Cao Y, Yan J. CN Patent CN115504886A 2022
Correspondence
Publication History
Received: 30 September 2025
Accepted after revision: 29 November 2025
Article published online:
20 January 2026
© 2026. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/).
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1a Yatav P, Kumar A, Althagafi I, Nemaysh V, Rai R, Pratap R. Curr Top Med Chem 2021; 21: 1587-1622
- 1b Faheem, Kumar BK, KVGC S, Chander S, Kunjiappan S, Murugesan S. RSC Adv 2021; 11: 12254-12287
- 1c Khadem S, Marles RJ. Nat Prod Res 2025; 399: 182-194
- 2a Ishihara Y, Kiyota Y, Goto G. Chem Pharm Bull 1990; 38: 3024-3030
- 2b Nishijima K, Shinkawa T, Ito M. et al. Eur J Med Chem 1998; 33: 763-774
- 3 Pearson WH, Fang WK. J Org Chem 2000; 65: 7158-7174
- 4a Hofmann F, Gartner C, Kretzschmar M, Schneider C. Synthesis 2022; 54: 1055-1080
- 4b Kretzschmar M, Hofmann F, Moock D, Schneider C. Angew Chem Int Ed 2018; 57: 4774-4778
- 5a Alicea J, Wolfe JP. J Org Chem 2014; 79: 4212-4217
- 5b Culberson MR, Dong S, Wolfe JP. Org Lett 2025; 27: 6445-6448
- 6 This work derives from and extends from the work described in Dr. Gabriel Gonzalez’s Ph.D. Dissertation: Gonzalez, Gabrial A., Ph.D. Dissertation, University of Michigan, 2024. https://www.proquest.com/dissertations-theses/studies-on-development-palladium-catalyzed-alkene/docview/3065166871/se-2
- 7 Wolfe JP. Top Heterocycl Chem 2013; 1-38
- 8 Wolfe JP, Rennels RA, Buchwald SL. Tetrahedron 1996; 52: 7525-7546
- 9a Wolfe JP, Wagaw S, Buchwald SL. J Am Chem Soc 1996; 118: 7215-7216
- 9b Wolfe JP, Buchwald SL. J Org Chem 2000; 65: 1144-1157
- 9c Schulummer B, Scholz U. Adv Synth Catal 2004; 346: 1599-1626
- 9d Muci AR, Buchwald SL. Top Curr Chem 2002; 219: 131-209
- 10 Netherton MR, Fu GC. Org Lett 2001; 3: 4295-4298
- 11 Schultz DM, Wolfe JP. Org Lett 2011; 13: 2962-2965
- 12a Grushin VV, Alper H. Chem Rev 1994; 94: 1047-1062
- 12b Portnoy M, Milstein D. Organometallics 1993; 12: 1665-1673
- 13 Tietze LF, Dufert MA, Hungerland T, Oum K, Lenzer T. Chem Eur J 2011; 17: 8452-8461
- 14 Cho SH, Hartwig JF. J Am Chem Soc 2013; 135: 8157-8160
- 15 Athelstan J, Beckwit L, Sendaba G. Aust J Chem 1992; 45: 289-308
- 16 Taber DF, Malcollm SC. J Org Chem 2001; 66: 944-953
- 17 Minatti A, Buchwald SL. Org Lett 2008; 10: 2721-2724
- 18 Zhang Z, Dwoskin LP, Crooks PA. Tetrahedron Lett 2011; 52: 2667-2669
- 19 Beyer TA, Chambers RJ, Lam K, Li M, Morrell AI, Thompson DD. WO Patent WO2005061497A1 2005
- 20 Xie Y, Zhang Q, Zhang H, Luo G, Cao Y, Yan J. CN Patent CN115504886A 2022
