
Subscribe to RSS
Please copy the URL and add it into your RSS Feed Reader.
https://www.thieme-connect.de/rss/thieme/en/10.1055-s-00000083.xml
Synlett 2013; 24(16): 2114-2118
DOI: 10.1055/s-0033-1339494
DOI: 10.1055/s-0033-1339494
letter
Site-Selective Suzuki–Miyaura Cross-Coupling Reactions of the Bis(triflate) of Methyl 3,7-Dihydroxy-2-naphthoate
Further Information
Publication History
Received: 07 June 2013
Accepted after revision: 09 July 2013
Publication Date:
14 August 2013 (online)
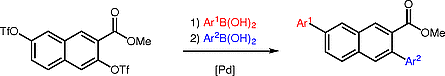

Abstract
Arylated naphthalenes were prepared by Suzuki–Miyaura reactions of the bis(triflate) of methyl 3,7-dihydroxy-2-naphthoate. The reactions proceeded with very good site-selectivity in favor of the sterically less hindered position 7, despite the proximity of position 3 to the electron-withdrawing ester group which plays a decisive role for the site-selectivity of Suzuki–Miyaura reactions of related substrates.
-
References and Notes
- 1a Donaldson N. The Chemistry and Technology of Naphthalene Compounds. Edward Arnold; London: 1958
- 1b Cullmann F, Schmidt A, Schuld F, Trennheuser ML, Becker H. Phytochemistry 1999; 52: 1647
- 1c Jiang Z.-H, Tanaka T, Inutsuka C, Kouno I. Chem. Pharm. Bull. 2001; 49: 737
- 1d Kongkathip N, Luangkamin S, Kongkathip B, Sangma C, Grigg R, Kongsaeree P, Prabpai S, Pradidphol N, Piyaviriyagul S, Siripong P. J. Med. Chem. 2004; 47: 4427
- 1e Ovenden SP. B, Yu J, Wan SS, Sberna G, Tait RM, Rhodes D, Cox S, Coates J, Walsh NG, Meurer-Grimes BM. Phytochemistry 2004; 65: 3255
- 1f Fedoreyev SA, Veselova MV, Krivoschekova OE, Mischenko NP, Denisenko VA, Dmitrenok PS, Glazunov VP, Bulgakov VP, Tchernoded GK, Zhuravlev YN. Planta Med. 2005; 71: 446
- 1g Bringmann G. The Naphthyl Isoquinoline Alkaloids, In The Alkaloids . Vol. 29. Brossi A. Academic Press; New York: 1986: 141
- 1h Bringmann G, Götz R, Keller PA, Walter R, Boyd MR, Lang F, Garcia A, Walsh JJ, Tellitu I, Bhaskar KV, Kelly TR. J. Org. Chem. 1998; 63: 1090
- 2a Hasegawa M, Takenouchi K, Takahashi K, Takeuchi T, Komoriya K, Uejima K. Microbiology 2007; 153: 2613
- 2b Silva O, Elsa T. J. Nat. Prod. 2003; 66: 447
- 2c De Koning CB, Michael JP, van Otterlo WA. L. Tetrahedron Lett. 1999; 40: 3037
- 2d Manfredi KP, Blunt JW, Cardellina JH. II, Macmahon JB, Pannell LK, Cragg GM, Boyd MR. J. Med. Chem. 1991; 34: 3402
- 2e Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Anticancer Res. 2004; 24: 2783
- 2f Signorelli P, Ghidoni R. J. Nutr. Biochem. 2005; 16: 449
- 2g Minutolo F, Sala G, Bagnacani A, Bertini S, Carboni I, Placanica G, Prota G, Rapposelli S, Sacchi N, Macchia M, Ghadoni R. J. Med. Chem. 2005; 48: 6783
- 2h Viswanathan GS, Wang M, Li CJ. Angew. Chem. Int. Ed. 2002; 41: 2138
- 2i Kozik B, Burgie ZJ, Sepiot JJ, Wilamowski J, Kuczynski M, Góra M. Environ. Biotechnol. 2006; 20
- 3a Kim JN, Im YJ, Gong JH, Lee KY. Tetrahedron Lett. 2001; 42: 4195
- 3b de Koning CB, Michael JP, Rousseau AL. Tetrahedron Lett. 1997; 38: 893
- 3c Viswanathan GS, Wang M, Li C.-J. Angew. Chem. Int. Ed. 2002; 41: 2138
- 3d Kabalka GW, Ju Y, Wu Z. J. Org. Chem. 2003; 68: 7915
- 3e Asao A, Takahashi K, Lee S, Kasahara T, Yamamoto Y. J. Am. Chem. Soc. 2002; 124: 12650
- 3f Asao A, Nogami T, Lee S, Yamamoto Y. J. Am. Chem. Soc. 2003; 125: 10921
- 3g Rengarajan B, Vanajakshi G. Org. Lett. 2009; 11: 3116
- 3h Saito S, Yamamoto Y. Chem. Rev. 2000; 100: 2901
-
3i Schore NE. Chem. Rev. 1988; 88: 1081
-
3j Lautens M, Klute W, Tam W. Chem. Rev. 1996; 96: 49
- 3k Grotjahn DB In Comprehensive Organometallic Chemistry II . Vol. 12. Hegedus LS, Abel EW, Stone FG. A, Wilkinson G. Pergamon Press; Oxford: 1995: 741
- 4a Schröter S, Stock C, Bach T. Tetrahedron 2005; 61: 2245
- 4b Schnürch M, Flasik R, Khan AF, Spina M, Mihovilovic MD, Stanetty P. Eur. J. Org. Chem. 2006; 3283
- 4c Rossi R, Bellina F, Lessi M. Adv. Synth. Catal. 2012; 354: 1181
- 5 For a simple guide to predict the regioselectivity of Pd catalyzed cross-coupling reactions, see: Handy ST, Zhang Y. Chem. Commun. 2006; 299
- 6 Ibad MF, Eleya N, Obaid-Ur-Rahman A, Mahal A, Hussain M, Villinger A, Langer P. Synthesis 2011; 2101
- 7 Methyl 3,7-Dihydroxy-2-naphthoate (2) To a solution of 3,7-dihydroxy-2-naphthoic acid (1, 2.0 g, 9.79 mmol) in DMF (30 mL), dimethyl sulfate (2.76 g, 21.93 mmol) and DIPEA (1.4 g, 10.76 mmol) were added. The reaction mixture was heated for 1 h at 85 °C. After cooling to r.t., the mixture was poured into ice water. A white precipitate formed which was filtered off, and the filtrate was concentrated in vacuo. The product 2 was isolated by column chromatography (flash silica gel, heptane–EtOAc) as a yellow solid (1.6 g, 77%); mp 101–102 °C. 1H NMR (300 MHz, CDCl3): δ = 3.95 (s, 3 H, OCH3), 4.79 (s, 1 H, OH), 7.04–7.05 (m, 1 H, ArH), 7.08 (dd, J = 2.4, 8.8 Hz, 1 H, ArH), 7.20 (s, 1 H, ArH), 7.55 (d, J = 8.9 Hz, 1 H, ArH), 8.25 (s, 1 H, ArH), 10.19 (s, 1 H, OH). 13C NMR (62.90 MHz, CDCl3): δ = 52.5 (OCH3), 110.1, 111.8 (CH), 114.6 (C), 121.6 (CH), 127.7 (C), 128.1, 130.3 (CH), 133.4, 151.7, 154.7 (C), 170.2 (CO). IR (KBr): ν = 3328 (m), 3038, 3003, 2953 (w), 1681 (m), 1651, 1633, 1609, 1576, 1556 (w), 1531 (m), 1505, 1479, 1462 (w), 1441, 1392, 1345, 1263, 1214 (s), 1180, 1147, 1130, 1072 (m), 1012, 969 (w), 944, 903, (w), 860 (s), 836, 812 (m), 783 (s), 746 (w), 716 (s), 665, 620, 610 (w), 587, 550 (w) cm–1. GC–MS (EI, 70 eV): m/z (%) = 219 (8) [M + H]+, 218 (52) [M]+, 187 (17), 186 (100), 185 (10), 159 (10), 158 (80), 130 (45), 102 (20). HRMS (EI, 70 eV): m/z calcd for C12H10O4 [M]+: 218.0579; found: 218.0599.
- 8 Methyl 3,7-Bis[4-(trifluoromethyl)sulfonyloxy-phenyl]-2-naphthoate (3) To a CH2Cl2 solution (46 mL) of 2 (1.0 g, 4.6 mmol) was added pyridine (1.5 mL, 18.4 mmol), and the solution was stirred at 20 °C for 10 min under an argon atmosphere. Then Tf2O (1.8 mL, 11.0 mmol) was added at –78 °C, and the reaction mixture was allowed to warm to r.t. and was stirred for 14 h. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The product 3 was isolated by column chromatography (flash silica gel, heptane–EtOAc) as a colorless solid (1.9 g, 87%); mp 75–77 °C. 1H NMR (300 MHz, CDCl3): δ = 4.06 (s, 3 H, OCH3), 7.47–7.70 (m, 1 H, ArH), 7.85 (s, 1 H, ArH), 7.94 (d, J = 2.3 Hz, 1 H, ArH), 8.03 (d, J = 9.0 Hz, 1 H, ArH), 8.72 (s, 1 H, ArH). 13C NMR (62.90 MHz, CDCl3): δ = 52.99 (OCH3), 117.7 (q, J F–C = 320.2 Hz, CF3), 117.9 (q, J F–C = 320.8 Hz, CF3), 120.4, 121.2, 123.6 (CH), 130.5 (C), 130.5 (CH), 131.8, 133.8 (C), 134.6 (CH), 145.5, 148.3 (C), 163.7 (CO). IR (KBr): ν = 3070, 2963 (w), 1713 (s), 1678, 1633 (w), 1599 (m), 1502, 1461, 1443 (w), 1426, 1398 (s), 1365, 1309 (m), 1276 (s), 1249 (m), 1203, 1131, 1116, 1048 (s), 965 (w), 937, 909, 937, 909, 845, 813, 795 (s), 779, 766, 754, 739 (m), 697 (w), 676 (m), 650, 609, (s), 597, 582 (m), 565 (w) cm–1. GC–MS (EI, 70 eV): m/z (%) = 483 (10) [M + H]+, 482 (61) [M]+, 451 (16), 350 (14), 349 (100), 257 (24), 129 (17). HRMS (ESI-TOF/MS): m/z calcd for C14H9F6O8S2 [M + H]+: 482.96375; found: 482.96348.
- 9 General Procedure for the Synthesis of 5a–j A THF solution of 3 (0.145 mmol), K3PO4 (1.5 equiv), Pd(PPh3)4 (3 mol%), and arylboronic acid 4 (1.2 equiv) was stirred at 20 °C for 9 h under argon atmosphere. To the reaction mixture were added H2O (20 mL) and CH2Cl2 (25 mL). The organic and the aqueous layers were separated, and the latter was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, heptane–EtOAc = 100:5).
- 10 Methyl 7-(3,5-Dimethylphenyl)-3-(trifluoro-methylsulfonyloxy)-2-naphthoate (5a) Starting with 3 (70 mg, 0.145 mmol), 4a (21 mg, 0.145 mmol), Pd(PPh3)4 (5 mg, 3 mol%, 0.0043 mmol), K3PO4 (30 mg, 0.145 mmol), and THF (4 mL), 5a was isolated as a white solid (45 mg, 70%), mp 102–104 °C. 1H NMR (300 MHz, CDCl3): δ = 2.35 (s, 6 H, 2CH3), 3.96 (s, 3 H, OCH3), 7.01 (s, 1 H, ArH), 7.24 (br s, 2 H, ArH), 7.68–8.08 (m, 4 H, ArH), 8.63 (s, 1 H, ArH). 19F NMR (282.4 MHz, CDCl3): δ = –73.1. 13C NMR (75.5 MHz, CDCl3): δ = 20.4 (2 CH3), 51.7 (OCH3), 117.8 (q, J C–F = 321.3, CF3), 120.8, 125.3, 126.6, 128.1, 129.7, 129.8 (CH), 131.8, 134.0 (C), 135.0 (CH), 138.7, 138.8 139.8, 141.7, 144.6 (C), 164.4 (C=O). IR (KBr,): ν = 3028, 3012, 2958, 2920, 2851 (w), 1721 (s), 1632 (w), 1596 (m), 1462 (w), 1420 (s), 1364, 1319, 1303, 1285, 1261, 1247 (w), 1198 (s), 1134, 1112, 1051, 1018, 946 (m), 919, 903, 886, 852 (w), 826, 807, 776, 764, 722 (s), 698, 667, 631, 612, 594, 574, 536 (m) cm–1. GC–MS (EI, 70eV): m/z (%) = 438 (44) [M+], 305 (100), 277 (8), 275 (15), 247 (39), 219 (20), 202 (24). HRMS (EI, 70 eV): m/z calcd for C21H17O5F3S [M]+: 438.07433; found: 438.07419.
- 11 CCDC 950296 (5g) and CCDC 950297 (6c) contain all crystallographic details of this publication and are available free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html or can be ordered from the following address: Cambridge Crystallographic Data Centre, 12 Union Road, GB-Cambridge CB21EZ; fax: +44(1223)336033; or deposit@ccdc.cam.ac.uk.
- 12 General Procedure for the Synthesis of 6a–i A solution of 3 (75 mg, 0.155 mmol), K3PO4 (3.0 equiv), Pd(PPh3)4 (6 mmol%), and arylboronic acid 3 (2.4 equiv) in 1,4-dioxane (3 mL) was stirred at 120 °C for 4 h under argon atmosphere. To the reaction mixture were added H2O (20 mL) and CH2Cl2 (25 mL). The organic and the aqueous layers were separated, and the latter was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, heptane–EtOAc).
- 13 Methyl 3,7-Bis(3,5-dimethylphenyl)-2-naphthoate (6a) Starting with 3 (75 mg, 0.155 mmol), 4a (47 mg, 0.31 mmol), Pd(PPh3)4 (10.7 mg, 6 mol%, 0.0093 mmol), K3PO4 (98.5 mg, 0.465 mmol), and 1,4-dioxane (3 mL), 6a was isolated as a white solid (50 mg, 83%), mp 175–176 °C. 1H NMR (300 MHz, CDCl3): δ = 2.28 (s, 6 H, 2CH3), 2.32 (s, 6 H, 2 CH3), 3.62 (s, 3 H, OCH3), 6.92 (br s, 1 H, ArH), 6.95 (br s, 3 H, ArH), 7.24 (br s, 2 H, ArH), 7.70–7.74 (m, 2 H, ArH), 7.78–7.80 (d, J = 8.6 Hz, 1 H, ArH), 7.99 (br s, 1 H, ArH), 8.29 (s, 1 H, ArH). 13C NMR (75.46 MHz, CDCl3): δ = 21.3, 21.4 (4 CH3), 52.0 (OCH3), 125.3, 126.1, 126.3, 128.0, 128.1, 128.8, 129.3 (CH), 129.7 (C), 130.9 (CH), 131.7, 133.4, 137.5, 138.4, 138.8, 139.5, 140.5, 141.2 (C), 169.2 (CO). IR (KBr): ν = 3453, 3026, 2986, 2944, 2914, 2857, 1935, 1841, 1787 (w), 1734 (s), 1693 (w), 1589 (m), 1496, 1454, 1443, 1430, 1397, 1375, 1462, 1325 (w), 1282 (m), 1268, 1258 (s), 1226, 1203, 1192, 1147 (m), 1100 (s), 1047 (m), 969, 943 (w), 922 (s), 897, 943 (w), 922, 897 (m), 852 (s), 833, 8220, 792 (m), 771, 760, 734, 721, 702 (w), 689 (s), 637, 622, 608, 574, 559, 543, 535 (w) cm–1. GC–MS (EI, 70 eV): m/z (%) = 394 (100) [M]+, 363 (13). HRMS (EI, 70 eV): m/z calcd for C28H26O2 [M]+: 394.19273; found: 394.192591.
- 14 Methyl 7-(4-Methoxyphenyl)-3-(4-tolyl)-2-naphthoate (7) The reaction was carried out in a pressure tube. To the THF suspension (4 mL) of 3 (70 mg, 0.145 mmol), arylboronic acid Ar1B(OH)2 4 (21 mg, 0.145 mmol), and Pd(PPh3)4 (3 mol%) was added K3PO4 (30 mg, 0.145 mmol), and the resulting solution was degassed by bubbling argon through the solution for 10 min. The mixture was stirred at 20 °C under an argon atmosphere for 9 h. Arylboronic acid Ar2B(OH)2 (0.035 mmol), Pd(PPh3)4 (3 mol%), and K3PO4 (30 mg, 0.145 mmol) were added. The reaction mixtures were heated under an argon atmosphere at 105 °C for 8 h. They were diluted with H2O and extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated in vacuo. The residue was purified by flash chromatography (silica gel, heptane–EtAOc) to give 7 as a white solid (55 mg, 85%), mp 82–83 °C. 1H NMR (300 MHz, CDCl3): δ = 2.36 (s, 3 H, CH3), 3.83 (s, 3 H, OCH3), 3.96 (s, 3 H, OCH3), 6.91 (d, 2 H, J = 8.8 Hz, ArH), 7.23–7.31 (m, 4 H, ArH), 7.56 (d, 2 H, J = 8.8 Hz, ArH), 7.74 (s, 1 H, ArH), 7.75–7.77 (m, 2 H, ArH), 7.82 (s, 1 H, ArH), 8.06 (s, 1 H, ArH). 13C NMR (75.5 MHz, CDCl3): δ = 20.1 (CH3), 52.7, 55.4 (OCH3), 113.1, 119.8 (CH), 121.3 (C), 125.3, 126.2, 126.7, 127.1, 128.5, 128.8 (CH), 130.8, 132.9 (C), 133.9 (CH), 134.2, 134.6, 135.8, 137.2, 139.9, 143.6 (C), 164.3 (C=O). FTIR (KBr): ν = 3002, 2954, 2927, 2847 (w), 1726 (s), 1632, 1599, 1509, 1488, 1459 (w), 1422 (s), 1402, 1370, 1314, 1286, 1263, 1249 (w), 1202, 1136 (s), 1052, 1031 (m), 969, 939 (w), 897, 853, 810, 792, 776, 722 (m), 696, 667, 643 (w), 609, 600 (m) cm–1. GC–MS (EI, 70eV): m/z (%) = 382 (87) [M+], 367 (9), 311 (100), 281 (23), 207 (50), 198 (18). HRMS (ESI-TOF): m/z calcd for C26H22O3 [M + H]+: 383.15678; found: 383.15623.
For reviews of cross-coupling reactions of polyhalogenated heterocycles and triflates, see: