

Subscribe to RSS
DOI: 10.1055/s-0036-1590978
Synthesis of N-Arylsulfonamides by a Copper-Catalyzed Reaction of Chloramine-T and Arylboronic Acids at Room Temperature
Authors
This work was supported by the National Natural Science Foundation of China (No. 21502069), the Science and Technology Project of the Education Department of Jiangxi Province (No. GJJ161236), and the Key Discipline Project of Nanchang Normal University (No. NSXK20141003).
Publication History
Received: 14 June 2017
Accepted after revision: 04 August 2017
Publication Date:
29 August 2017 (online)

Abstract
A copper-catalyzed Chan–Lam-coupling-like reaction of a (het)arylboronic acid and chloramine-T (or a related compound) has been developed for the synthesis of N-arylsulfonamides at room temperature in moderate to good yields, with good tolerance of functional groups. In this process, it is believed that chloramine-T serves as an electrophile.
Chloramine-T {sodium chloro[(4-methyl phenyl)sulfonyl]azanide} is recognized as an important source of N-building blocks (especially of nitrenes), because of its ready commercial availability.[1] Chlorosulfamidation, aminohydroxylation, or aziridination of olefins and sulfamidation of relatively highly reactive alkanes can be performed by using chloroamine-T with or without metal catalysis to give chlorosulfamidated products [Scheme [1, a]],[2] N-protected β-amino alcohols [Scheme [1, b]],[3] aziridines [Scheme [1, c]],[4] or sulfamidated compounds [Scheme [1, d]],[5] respectively. In these procedures, nitrene species derived from chloramine-T by metal- or nonmetal-assisted removal of sodium chloride are generally regarded as intermediates. In 1998, Smith and co-workers discovered that chloramine-T reacts with alkylboranes in the absence of a catalyst to give N-alkylsulfonamides.[6] Recently, Chandrasekaran and co-workers found that chloramine-T behaves as an efficient dual nucleophile when vinylcyclopropanes are used as reaction partners in the presence of bromine, leading to [4.4.0] and [4.3.0] bicyclic amidines through tandem ring opening of the vinylcyclopropane and double-nucleophilic addition of chloramine-T; their results also showed that chloramine-T can be regarded as a dual nucleophile when its chlorine atom is removed in the form of BrCl.[7] Theoretically, chloramine-T can also serve as an electrophile, because it readily undergoes oxidative addition at the N–Cl bond in the presence of an appropriate metal catalyst.[8] As such, we were interested in exploring this electrophilic reactivity of chloramine-T for the synthesis of pharmaceutically interesting motifs.
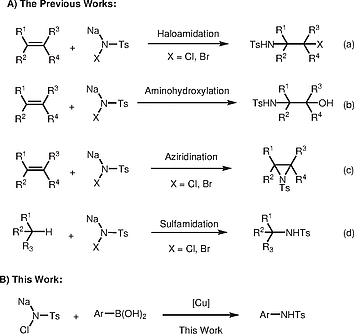
The N-arylsulfonamide group is an important moiety in medicinal chemistry because of its presence in many useful molecules with potential bioactivities.[9] Consequently, tremendous efforts have been made to develop methods for its synthesis. N-Arylsulfonamides are normally prepared by nucleophilic substitution of a sulfonyl chloride with an aromatic amine. Although effective, this classical approach requires the use of mutagenic sulfonyl chlorides and genotoxic aryl amines. Alternatively, the metal-catalyzed cross-coupling of sulfonamides with aromatic halides or other analogous compounds permits the synthesis of N-arylsulfonamide without the use of sulfonyl chloride, but these reactions are generally require harsh conditions because of the low nucleophilicity of sulfonamides.[10] In 1998, Chan and co-workers reported a synthesis of N-arylsulfonamides from sulfonamides and arylboronic acids.[11] This C–N bond-formation reaction can be performed under mild conditions; however, it suffers from the drawback of requiring the use of a stoichiometric amount of Cu(OAc)2. Since this reaction was first reported, many modifications have been demonstrated. For instance, Lam et al. developed several catalytic copper/oxidant systems to carry out the N-arylation of NH-containing substrates.[12] Although catalytic, those conditions work only for a limited range of sulfonamides. In 2014, Nasrollahzadeh and co-workers developed a simple Cu(OAc)2-catalyzed arylation of sulfonamides in water under ligand-free conditions, but this transformation requires elevated temperatures and is incompatible with ortho-substituted arylboronic acids.[13] Another elegant example, developed by Kim and co-workers, involved a copper-catalyzed Chan–Lam cross-coupling between an arylboronic acid and a sulfonyl azide to give various N-arylsulfonamides.[14] However, in general, sulfonyl azides are not shelf-stable and they require precautions in handling. Therefore, it is desirable to develop more-efficient and more-convenient methods for the synthesis of N-arylsulfonamides.
a Reagents and conditions: chloramine-T (0.3 mmol ), 4-TolB(OH)2 (1.2 equiv), base (1.0 equiv), catalyst (0.05 equiv), solvent (1.5 mL), 6 h.
b Isolated yield.
c Reaction time 12 h.
d Reaction time 24 h.
e 4-TolB(OH)2 (2.0 equiv).
f t-BuOK (1.5 equiv).
g t-BuOK (2.0 equiv).
Inspired by the methods described above, we decided to examine the preparation of N-arylsulfonamides from chloramine-T. Initially, we examined the reaction of 4-tolylboronic acid (1k) with chloramine-T (2a) as a model reaction for the optimization of the conditions. To our delight, the desired 4-methyl-N-(4-tolyl)benzenesulfonamide (3k) was obtained in the presence of 5 mol% of Cu(OAc)2 and 1.0 equivalent of NaOH in MeOH as a solvent at 60 °C (Table [1], entry 1). Encouraged by this positive result, we examined various other factors (Table [1]). From a screening of bases, it emerged that potassium tert-butoxide was the optimal choice, providing the desired N-arylsulfonamide 3k in 42% isolated yield (Table [1], entry 4); other bases (triethylamine, potassium hydroxide, and DBU) gave inferior yields (Table [1], entries 2, 3, and 5). Subsequently, various copper sources were also examined and copper (II) catalysts were found to be more efficient than copper (I) catalysts for the formation of 3a, and no better copper source than Cu(OAc)2 was found (Table [1], entries 6–11). We also examined the effect of the solvent, and found that protic solvents such as ethanol, isopropyl alcohol, and tert-butanol tended to give better results than did aprotic solvents such as methyl tert-butyl ether, tetrahydrofuran, 1,2-dichloroethane, or acetonitrile (Table [1], entries 12–18). Among the protic solvents, ethanol gave the best yield (54%) of the N-arylsulfonamide 3a (Table [1], entry 12). A subsequent evaluation of the effect of the temperature showed that reducing the reaction temperature to room temperature had no significant effect on the yield (Table [1], entry 19). The yield increased slightly when the reaction time was prolonged to 12 hours (Table [1], entry 20). A further increase in the reaction time or an increased loading of the arylboronic acid did not improve the yield (Table [1], entries 21 and 22, respectively). Interestingly, when the amount of the potassium tert-butoxide base was increased to 1.5 equivalents, the yield improved to 60% (Table [1], entry 23). A further increase in the amount of base did not produce any improvement (Table [1], entry 24).
With the optimized conditions in hand [Cu(OAc)2 (5 mol%) and t-BuOK (1.5 equiv) in EtOH at room temperature], we examined the scope and generality of the reaction of various arylboronic acids 1 with chloramine-T (2a) (Scheme [2]), and obtained the expected series of N-arylsulfonamides 3. To our delight, arylboronic acids with electron-withdrawing substituents, such as fluoro or trifluoromethyl groups, were compatible for the reaction, and gave the corresponding N-arylsulfonamides 3b–j in moderate yields. For example, the reaction of [4-(trifluoromethyl)phenyl]boronic acid and chloramine-T (2a) gave the N-arylsulfonamide 3e in 51% yield. Moreover, a bromo substituent survived the reaction; the reaction of (4-bromophenyl)boronic acid (1, R1 = 4-Br) or (2-bromophenyl)boronic acid (1, R1 = 2-Br) with chloramine-T (2a) gave N-arylsulfonamides 3i and 3j efficiently in 67 and 53% yield, respectively, under the standard conditions. Furthermore, when (3,5-dichlorophenyl)boronic acid was used as a substrate, the desired N-arylsulfonamide 3h was obtained in 53% isolated yield.

Arylboronic acids with electron-donating substituents on the aryl moiety also reacted efficiently with chloramine-T (2a), giving the corresponding N-arylsulfonamide 3k–p. For example, the reaction of 3-tolylboronic acid (1, R1 = 3-Me) and (2-methoxyphenyl)boronic acid (1, R1 = 2-OMe) gave the N-arylsulfonamides 3l and 3o in 72 and 73% yield, respectively. 1-Naphthylboronic acid and 2-naphthylboronic acid were also suitable reactants, providing the corresponding N-arylsulfonamides 3r and 3s in 49 and 69% yields, respectively. However, 1,3-benzodioxol-5-ylboronic acid was a less efficient reactant in the synthesis of the N-arylsulfonamide 3q (37% yield), probably due to its high reducibility and reactivity for homocoupling. Additionally, hetarylboronic acids were good reaction partners for the formation of corresponding N-hetarylsulfonamides. For instance, the reaction of [6-(dimethylamino)pyridin-3-yl]boronic acid gave the desired N-pyridinylsulfonamide 3t in 43% yield.
Sodium chloro[(4-chlorophenyl)sulfonyl]azanide (2b), an analogue of chloramine-T, was also tested for the synthesis of N-arylsulfonamides 4a–i (Scheme [2]). Azanide 2b was normally less efficient than chloramine-T in its reaction with arylboronic acid. For example, the reaction of compound 2b with (4-bromophenyl)boronic acid gave the desired N-arylsulfonamide 4h in 43% yield, whereas the corresponding reaction of chloramine-T gave a 67% yield of 3i. Additionally, 1-naphthylboronic acid was a good reaction partner for the synthesis of the N-arylsulfonamide 4i in 57% yield. To our surprise, sodium chloro(alkylsulfonyl)azanides were not compatible with the reaction (results not shown).
Although the precise mechanism of the sulfamidation reaction still remains uncertain, a possible reaction pathway is proposed on the basis of previous studies (Scheme [3]). Initially, transmetalation of the aryl group of the arylboronic acid with copper(II) and a subsequent ligand exchange with EtOK gives rise to species A, which is oxidized by a second equivalent of Cu(II), to form Cu(I) and the intermediate B; this is followed by C–O reductive elimination to provide Cu(I) and byproduct C.[15] Indeed, a trace of ethoxybenzene was observed by GC/MS analysis when the reaction of chloramine-T (2a) with 4-tolylboronic acid (1k) was carried out under the standard conditions. Subsequently, species D is produced through oxidative addition of Cu(I) to the N–Cl bond of chloramine-T; subsequent transmetalation of the arylboronic acid generates intermediate E.[8] Finally, a C–N reductive elimination of intermediate E delivers the desired N-arylsulfonamide and reforms the Cu(I) catalyst.[8a]

To demonstrate the potential synthetic application of the present method, we attempted a sulfamidation of (4-bromophenyl)boronic acid (1i) on a gram scale, and we obtained the desired product 3i in 61% yield (Scheme [4]).

In conclusion, we have developed a novel copper-catalyzed Chan–Lam-coupling-like reaction of arylboronic acids with sodium chloro(arylsulfonyl)azanides to give the corresponding N-arylsulfonamides at room temperature and in moderate to good yields with a good tolerance of functional groups.[16] In this process, we believe that the sodium chloro(arylsulfonyl)azanide serves as an electrophile (in a similar manner to halide) to oxidize the copper catalyst through oxidative addition. Exploration of chloramine-based chemistry for the synthesis of other bioactive motifs is ongoing in our laboratory. The results will be published in due course.
Acknowledgment
We are grateful to the Analysis and Testing Center of Jiangxi Normal University for the analytical data.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/s-0036-1590978.
- Supporting Information (PDF) (opens in new window)
-
References and Notes
- 1a Agnihotri G. Synlett 2005; 2857
- 1b Minakata S. Acc. Chem. Res. 2009; 42: 1172
- 1c Li Z. Capretto DA. He C. In Silver in Organic Chemistry . Harmata M. Wiley; Oxford: 2010. Chap. 6, 16
- 1d Goehring RR. In Handbook of Reagents for Organic Synthesis: Catalytic Oxidation Reagents . Fuchs PL. Wiley; Chichester: 2013: 142
- 2a Minakata S. Yoneda Y. Oderaotoshi Y. Komatsu M. Org. Lett. 2006; 8: 967
- 2b Wu X.-L. Wang G.-W. Eur. J. Org. Chem. 2008; 2008: 6239
- 2c Hayakawa J. Kuzuhara M. Minakata S. Org. Biomol. Chem. 2010; 8: 1424
- 2d Martínez C. Muñiz K. Adv. Synth. Catal. 2014; 356: 205
- 3a Li G. Chang H.-T. Sharpless KB. Angew. Chem. Int. Ed. 1996; 35: 451
- 3b Rubin AE. Sharpless KB. Angew. Chem. Int. Ed. 1997; 36: 2637
- 3c Sugimoto H. Mikami A. Kai K. Sajith PK. Shiota Y. Yoshizawa K. Asano K. Suzuki T. Itoh S. Inorg. Chem. 2015; 54: 7073
- 4a Ando T. Minakata S. Ryu I. Komatsu M. Tetrahedron Lett. 1998; 39: 309
- 4b Ando T. Kano D. Minakata S. Ryu I. Komatsu M. Tetrahedron 1998; 54: 13485
- 4c Jeong JU. Tao B. Sagasser I. Henniges H. Sharpless KB. J. Am. Chem. Soc. 1998; 120: 6844
- 4d Antunes AM. M. Marto SJ. L. Branco PS. Prabhakar S. Lobo AM. Chem. Commun. 2001; 405
- 4e Kano D. Minakata S. Komatsu M. J. Chem. Soc., Perkin Trans. 1 2001; 3186
- 4f Kumar GD. Baskaran S. Chem. Commun. 2004; 1026
- 4g Vyas R. Gao G.-Y. Harden JD. Zhang XP. Org. Lett. 2004; 6: 1907
- 4h Minakata S. Kano D. Oderaotoshi Y. Komatsu M. Angew. Chem. Int. Ed. 2004; 43: 79
- 4i Gao G.-Y. Harden JD. Zhang XP. Org. Lett. 2005; 7: 3191
- 5a Albone DP. Aujla PS. Taylor PC. J. Org. Chem. 1998; 63: 9569
- 5b Albone DP. Challenger S. Derrick AM. Fillery SM. Irwin JL. Parsons CM. Takada H. Taylor PC. Wilson DJ. Org. Biomol. Chem. 2005; 3: 107
- 5c Baumann T. Bachle M. Brase S. Org. Lett. 2006; 8: 3797
- 5d Bhuyan R. Nicholas KM. Org. Lett. 2007; 9: 3957
- 5e Harden JD. Ruppel JV. Gao G.-Y. Zhang XP. Chem. Commun. 2007; 4644
- 5f Takeda Y. Hayakawa J. Yano K. Minakata S. Chem. Lett. 2012; 41: 1672
- 6 Jigajinni VB. Pelter A. Smith K. Tetrahedron Lett. 1978; 19: 181
- 7a Minakata S. Kano D. Oderaotoshi Y. Komatsu M. Org. Lett. 2002; 4: 2097
- 7b Ganesh V. Sureshkumar D. Chanda D. Chandrasekaran S. Chem. Eur. J. 2012; 18: 12498
- 8a Kawano T. Hirano K. Satoh T. Miura M. J. Am. Chem. Soc. 2010; 132: 6900
- 8b Liu X.-Y. Gao P. Shen Y.-W. Lianga Y.-M. Adv. Synth. Catal. 2011; 353: 3157
- 9a Scozzafava A. Owa T. Mastrolorenzo A. Supuran CT. Curr. Med. Chem. 2003; 10: 925
- 9b Eschenburg S. Priestman MA. Abdul-Latif FA. Delachaume C. Fassy F. Schönbrunn E. J. Biol. Chem. 2005; 280: 14070
- 9c Stanton MG. Stauffer SR. Gregro AR. Steinbeiser M. Nantermet P. Sankaranarayanan S. Price EA. Wu G. Crouthamel M.-C. Ellis J. Lai M.-T. Espeseth AS. Shi X.-P. Jin L. Colussi D. Pietrak B. Huang Q. Xu M. Simon AJ. Graham SL. Vacca JP. Selnick H. J. Med. Chem. 2007; 50: 3431
- 9d Basanagouda M. Shivashankar K. Kulkarni MV. Rasal VP. Patel H. Mutha SS. Mohite AA. Eur. J. Med. Chem. 2010; 45: 1151
- 9e Chohan ZH. Youssoufi MH. Jarrahpour A. Ben Hadda T. Eur. J. Med. Chem. 2010; 45: 1189
- 10a Ley SV. Thomas AW. Angew. Chem. Int. Ed. 2003; 42: 5400
- 10b Ma D. Cai Q. Acc. Chem. Res. 2008; 41: 1450
- 10c Surry DS. Buchwald SL. Angew. Chem. Int. Ed. 2008; 47: 6338
- 10d Monnier F. Taillefer M. Angew. Chem. Int. Ed. 2009; 48: 6954
- 10e Qiao J. Lam P. Synthesis 2011; 829
- 10f Louillat ML. Patureau FW. Chem. Soc. Rev. 2014; 43: 901
- 11 Chan DM. T. Monaco KL. Wang R.-P. Winters MP. Tetrahedron Lett. 1998; 39: 2933
- 12 Lam PY. S. Vincent G. Clark CG. Deudon S. Jadhav PK. Tetrahedron Lett. 2001; 42: 3415
- 13 Nasrollahzadeh M. Ehsani A. Maham M. Synlett 2014; 25: 505
- 14 Moon SY. Nam J. Rathwell K. Kim W.-S. Org. Lett. 2014; 16: 338
- 15a King AE. Brunold TC. Stahl SS. J. Am. Chem. Soc. 2009; 131: 5044
- 15b King AE. Huffman LM. Casitas A. Costas M. Ribas X. Stahl SS. J. Am. Chem. Soc. 2010; 132: 12068
- 15c Rao DN. Rasheed S. Kumar KA. Reddy AS. Das P. Adv. Synth. Catal. 2016; 358: 2126
- 16 Reaction of Chloramines 2 with Arylboronic Acids 1; General Procedure A test tube equipped with a stirrer bar was charged with the appropriate chloramine 2 (0.3 mmol), arylboronic acid 1 (0.36 mmol), and t-BuOK (50.5 mg, 0.45 mmol). A solution of Cu(OAc)2 (2.7 mg, 0.015 mmol) in EtOH (1.5 mL) was then added, and the mixture was stirred under air at RT for 12 h. The heterogeneous mixture was then diluted with EtOAc (1 mL), and the resulting mixture was filtered through a pad of silica gel, which was washed with EtOAc (3 mL). The organic solutions were combined, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (silica gel, PE–EtOAc). 4-Methyl-N-phenylbenzenesulfonamide (3a) White solid; yield: 46 mg (62%); mp 100–101 °C. 1H NMR (400 MHz, CDCl3): δ = 7.67 (d, J = 8.3 Hz, 2 H), 7.22 (t, J = 7.9 Hz, 4 H), 7.09 (dd, J = 9.4, 8.3 Hz, 3 H), 6.97 (s, 1 H), 2.37 (s, 3 H). 13C NMR (101 MHz, CDCl3): δ = 143.87, 136.58, 136.14, 129.65, 129.30, 127.29, 125.30, 121.57, 21.52.
-
References and Notes
- 1a Agnihotri G. Synlett 2005; 2857
- 1b Minakata S. Acc. Chem. Res. 2009; 42: 1172
- 1c Li Z. Capretto DA. He C. In Silver in Organic Chemistry . Harmata M. Wiley; Oxford: 2010. Chap. 6, 16
- 1d Goehring RR. In Handbook of Reagents for Organic Synthesis: Catalytic Oxidation Reagents . Fuchs PL. Wiley; Chichester: 2013: 142
- 2a Minakata S. Yoneda Y. Oderaotoshi Y. Komatsu M. Org. Lett. 2006; 8: 967
- 2b Wu X.-L. Wang G.-W. Eur. J. Org. Chem. 2008; 2008: 6239
- 2c Hayakawa J. Kuzuhara M. Minakata S. Org. Biomol. Chem. 2010; 8: 1424
- 2d Martínez C. Muñiz K. Adv. Synth. Catal. 2014; 356: 205
- 3a Li G. Chang H.-T. Sharpless KB. Angew. Chem. Int. Ed. 1996; 35: 451
- 3b Rubin AE. Sharpless KB. Angew. Chem. Int. Ed. 1997; 36: 2637
- 3c Sugimoto H. Mikami A. Kai K. Sajith PK. Shiota Y. Yoshizawa K. Asano K. Suzuki T. Itoh S. Inorg. Chem. 2015; 54: 7073
- 4a Ando T. Minakata S. Ryu I. Komatsu M. Tetrahedron Lett. 1998; 39: 309
- 4b Ando T. Kano D. Minakata S. Ryu I. Komatsu M. Tetrahedron 1998; 54: 13485
- 4c Jeong JU. Tao B. Sagasser I. Henniges H. Sharpless KB. J. Am. Chem. Soc. 1998; 120: 6844
- 4d Antunes AM. M. Marto SJ. L. Branco PS. Prabhakar S. Lobo AM. Chem. Commun. 2001; 405
- 4e Kano D. Minakata S. Komatsu M. J. Chem. Soc., Perkin Trans. 1 2001; 3186
- 4f Kumar GD. Baskaran S. Chem. Commun. 2004; 1026
- 4g Vyas R. Gao G.-Y. Harden JD. Zhang XP. Org. Lett. 2004; 6: 1907
- 4h Minakata S. Kano D. Oderaotoshi Y. Komatsu M. Angew. Chem. Int. Ed. 2004; 43: 79
- 4i Gao G.-Y. Harden JD. Zhang XP. Org. Lett. 2005; 7: 3191
- 5a Albone DP. Aujla PS. Taylor PC. J. Org. Chem. 1998; 63: 9569
- 5b Albone DP. Challenger S. Derrick AM. Fillery SM. Irwin JL. Parsons CM. Takada H. Taylor PC. Wilson DJ. Org. Biomol. Chem. 2005; 3: 107
- 5c Baumann T. Bachle M. Brase S. Org. Lett. 2006; 8: 3797
- 5d Bhuyan R. Nicholas KM. Org. Lett. 2007; 9: 3957
- 5e Harden JD. Ruppel JV. Gao G.-Y. Zhang XP. Chem. Commun. 2007; 4644
- 5f Takeda Y. Hayakawa J. Yano K. Minakata S. Chem. Lett. 2012; 41: 1672
- 6 Jigajinni VB. Pelter A. Smith K. Tetrahedron Lett. 1978; 19: 181
- 7a Minakata S. Kano D. Oderaotoshi Y. Komatsu M. Org. Lett. 2002; 4: 2097
- 7b Ganesh V. Sureshkumar D. Chanda D. Chandrasekaran S. Chem. Eur. J. 2012; 18: 12498
- 8a Kawano T. Hirano K. Satoh T. Miura M. J. Am. Chem. Soc. 2010; 132: 6900
- 8b Liu X.-Y. Gao P. Shen Y.-W. Lianga Y.-M. Adv. Synth. Catal. 2011; 353: 3157
- 9a Scozzafava A. Owa T. Mastrolorenzo A. Supuran CT. Curr. Med. Chem. 2003; 10: 925
- 9b Eschenburg S. Priestman MA. Abdul-Latif FA. Delachaume C. Fassy F. Schönbrunn E. J. Biol. Chem. 2005; 280: 14070
- 9c Stanton MG. Stauffer SR. Gregro AR. Steinbeiser M. Nantermet P. Sankaranarayanan S. Price EA. Wu G. Crouthamel M.-C. Ellis J. Lai M.-T. Espeseth AS. Shi X.-P. Jin L. Colussi D. Pietrak B. Huang Q. Xu M. Simon AJ. Graham SL. Vacca JP. Selnick H. J. Med. Chem. 2007; 50: 3431
- 9d Basanagouda M. Shivashankar K. Kulkarni MV. Rasal VP. Patel H. Mutha SS. Mohite AA. Eur. J. Med. Chem. 2010; 45: 1151
- 9e Chohan ZH. Youssoufi MH. Jarrahpour A. Ben Hadda T. Eur. J. Med. Chem. 2010; 45: 1189
- 10a Ley SV. Thomas AW. Angew. Chem. Int. Ed. 2003; 42: 5400
- 10b Ma D. Cai Q. Acc. Chem. Res. 2008; 41: 1450
- 10c Surry DS. Buchwald SL. Angew. Chem. Int. Ed. 2008; 47: 6338
- 10d Monnier F. Taillefer M. Angew. Chem. Int. Ed. 2009; 48: 6954
- 10e Qiao J. Lam P. Synthesis 2011; 829
- 10f Louillat ML. Patureau FW. Chem. Soc. Rev. 2014; 43: 901
- 11 Chan DM. T. Monaco KL. Wang R.-P. Winters MP. Tetrahedron Lett. 1998; 39: 2933
- 12 Lam PY. S. Vincent G. Clark CG. Deudon S. Jadhav PK. Tetrahedron Lett. 2001; 42: 3415
- 13 Nasrollahzadeh M. Ehsani A. Maham M. Synlett 2014; 25: 505
- 14 Moon SY. Nam J. Rathwell K. Kim W.-S. Org. Lett. 2014; 16: 338
- 15a King AE. Brunold TC. Stahl SS. J. Am. Chem. Soc. 2009; 131: 5044
- 15b King AE. Huffman LM. Casitas A. Costas M. Ribas X. Stahl SS. J. Am. Chem. Soc. 2010; 132: 12068
- 15c Rao DN. Rasheed S. Kumar KA. Reddy AS. Das P. Adv. Synth. Catal. 2016; 358: 2126
- 16 Reaction of Chloramines 2 with Arylboronic Acids 1; General Procedure A test tube equipped with a stirrer bar was charged with the appropriate chloramine 2 (0.3 mmol), arylboronic acid 1 (0.36 mmol), and t-BuOK (50.5 mg, 0.45 mmol). A solution of Cu(OAc)2 (2.7 mg, 0.015 mmol) in EtOH (1.5 mL) was then added, and the mixture was stirred under air at RT for 12 h. The heterogeneous mixture was then diluted with EtOAc (1 mL), and the resulting mixture was filtered through a pad of silica gel, which was washed with EtOAc (3 mL). The organic solutions were combined, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (silica gel, PE–EtOAc). 4-Methyl-N-phenylbenzenesulfonamide (3a) White solid; yield: 46 mg (62%); mp 100–101 °C. 1H NMR (400 MHz, CDCl3): δ = 7.67 (d, J = 8.3 Hz, 2 H), 7.22 (t, J = 7.9 Hz, 4 H), 7.09 (dd, J = 9.4, 8.3 Hz, 3 H), 6.97 (s, 1 H), 2.37 (s, 3 H). 13C NMR (101 MHz, CDCl3): δ = 143.87, 136.58, 136.14, 129.65, 129.30, 127.29, 125.30, 121.57, 21.52.
