

Subscribe to RSS
DOI: 10.1055/s-0040-1706023
Recent Advances in Transition-Metal-Free (4+3)-Annulations
Authors
The authors would like to thank the Natural Sciences and Engineering Research Council (NSERC), Alphora Inc., and the University of Toronto for funding. H.L. thanks the NSERC Postgraduate Scholarships –Doctoral (PGS-D) program. X.A.-S. thanks the Ontario Graduate Scholarship (OGS) and the NSERC Canada Graduate Scholarships – Master’s (CGS-M) program. M.F.K. thanks the Deutscher Akademischer Austauschdienst (DAAD) (German Academic Exchange Service) and the Bayerischen Staat (State of Bavaria) for the PROSA LMU program.

Abstract
(4+3)-Annulations are incredibly versatile reactions which combine a 4-atom synthon and a 3-atom synthon to form both 7-membered carbocycles as well as heterocycles. We have previously reviewed transition-metal-catalyzed (4+3)-annulations. In this review, we will cover examples involving bases, NHCs, phosphines, Lewis and Brønsted acids as well as some rare examples of boronic acid catalysis and photocatalysis. In analogy to our previous review, we exclude annulations involving cyclic dienes like furan, pyrrole, cyclohexadiene or cyclopentadiene, as Chiu, Harmata, Fernándes and others have recently published reviews encompassing such substrates. We will however discuss the recent additions (2010–2020) to the literature on (4+3)-annulations involving other types of 4-atom-synthons.
1 Introduction
2 Bases
3 Annulations Using N-Heterocyclic Carbenes
3.1 N-Heterocyclic Carbenes (NHCs)
3.2 N-Heterocyclic Carbenes and Base Dual-Activation
4 Phosphines
5 Acids
5.1 Lewis Acids
5.2 Brønsted Acids
6 Boronic Acid Catalysis and Photocatalysis
7 Conclusion
Key words
annulation - (4+3)-annulations - organocatalysis - Lewis acids - Brønsted acids - phosphines - N-heterocyclic carbenesBiographical Sketches

Heather Lam was born in Toronto, Ontario, Canada. She is a Ph.D. graduate from Professor Mark Lautens’ group at the University of Toronto. She completed her B.Sc. in a Specialist Human Biology Program at the University of Toronto Scarborough in 2014. Between her graduation and starting in Professor Lautens’ group, she conducted research in the groups of Professor Xiao-an Zhang, synthesizing enzyme-activatable MRI contrast agents, and of Professor Mitchell Winnik, synthesizing a modified DFO compound for chelating radioactive 89Zr. Her research interests reside in dual catalysis and multicatalytic reactions and their application for the synthesis of small molecules. She is currently at Princeton University pursuing her post-doctoral studies under the guidance of Professor Todd Hyster in the area of biocatalysis and photocatalysis.

Xavier Abel-Snape was born in Hong Kong, HKSAR. He is currently an NSERC-funded 2nd year Ph.D. student in Professor Mark Lautens’ group at the University of Toronto. He completed his B.Sc. in chemistry at Université de Montréal, where he did two internships under the supervision of Professor Shawn K. Collins. His work comprised catalyzing cyclization reactions using UV light and iron-based complexes in batch and continuous flow set-ups as well as optimizing the synthesis and diversification of macrocyclic alkynyl sulfide peptides. He also conducted research at the Fraunhofer Institute for Microengineering and Microsystems in Mainz, Germany, where he worked on the synthesis of silica- and noble-metal-based nanoparticles using different microfluidic technologies. His research interests reside in transition-metal-catalyzed domino C–H activation reactions, such as Catellani chemistry.

Martin F. Köllen was born in Frankfurt am Main, Germany. He obtained his B.Sc. in chemistry and biochemistry at the Ludwig-Maximilians-Universität München in 2019, with a bachelor thesis in the group of Dr. Dorian Didier on single-pot access to bisorganoborinates and their applications in Zweifel-olefination. He is currently a master’s student at LMU München, majoring in organic and inorganic chemistry. During his master’s studies, he conducted research in the groups of Professor Franz Bracher, working on a total synthesis of an antiplasmodial alkaloid, and of Professor Wolfgang Schnick, synthesizing novel nitridosilicates with luminescent properties for use in LEDs. He also went abroad for a research internship in the group of Professor Mark Lautens at the University of Toronto, performing research on palladium-catalyzed domino-Heck/C–H activation and nickel-catalyzed carboiodination reactions.

Mark Lautens was born in Hamilton, Ontario, Canada. He obtained his B.Sc. degree from the University of Guelph followed by doctoral studies at the University of Wisconsin–Madison under the direction of Professor Barry M. Trost where he discovered Mo-catalyzed allylic alkylation and the Pd-catalyzed enyne cycloisomerization. In 1985, he moved to Harvard University where he conducted his NSERC post-doctoral fellowship with Professor David A. Evans on studies directed towards the synthesis of bryostatin. He joined the University of Toronto in 1987 as an NSERC University Research Fellow and Assistant Professor. Since 1998 he has held an Endowed Chair, the AstraZeneca Professor of Organic Synthesis, and from 2003–2013 he was named an NSERC/Merck Frosst Industrial Research Chair in New Medicinal Agents via Catalytic Reactions. In 2012, he was appointed as University Professor, the highest rank at the University of Toronto. Most recently he was awarded Officer of the Order of Canada, the highest civilian honor in Canada. His current research interests are in multicatalyst reactions, isomerization reactions, and applications of catalysis in the synthesis of bioactive compounds.
Introduction
Methods for the synthesis of medium-sized rings continue to be of interest to the organic chemistry community. In the past decade, there have been a multitude of applications of (4+3)-annulations towards the formation of 7-membered rings (Figure 1.1), which are encountered in therapeutic agents and natural products. Many of these reactions involve the use of cyclic diene systems such as furans, pyrroles, cyclopentadienes and cyclohexadienes.[1]

In the literature, the terms annulation and cycloaddition are often used interchangeably. By IUPAC definition, cycloadditions involve two or more unsaturated molecules (or parts of the same molecule), which combine to form a cyclic adduct with a net reduction in bond multiplicity. In many of the examples featured in this review, although a cyclic adduct is formed, a net reduction of bond multiplicity does not always occur. Hence, the term that will be used to describe the reactions in this review will be annulation rather than cycloaddition.
Harmata has published multiple reviews on the applicability of (4+3)-annulations between allylic cations and cyclic dienes.[2] [3] Similarly, Fernández published an in-depth review on intramolecular transition-metal (PtII and AuI salts) catalyzed (4+3)-annulations of allylic cations and dienes in 2012.[4] This review discussed mechanistic proposals and transition states of (4+3)-annulations. A recent review from Chiu outlined the utility of (4+3)-annulations in the synthesis of natural products.[5] Swamy also presented a review involving the synthesis of 7-membered heterocycles and carbocycles in 2020.[6] Consequently, our review will cover examples involving acyclic 4-atom equivalents in literature in recent years (2010–2020). Following our previous discussion of transition-metal-catalyzed (4+3)-annulations, we will now discuss (4+3)-annulations involving bases, N-heterocyclic carbenes (NHCs), phosphines, Lewis acids, Brønsted acids, and those involving dual-activation modes.[7] A wide range of substrates are covered in this review (Figure 1.2). Some coupling partners are bench stable and are added directly to the reaction at the outset. Others can be generated during the reaction and generally require a catalyst for their formation. Due to the compatibility of bases with NHCs and phosphines, this review will also cover the examples of dual-activation modes.

2
Bases
The following examples employ stoichiometric amounts of an inorganic Brønsted base to drive the reaction to product. As such, the general role of a base in these reactions is to deprotonate one of the 4- or 3-atom synthons. This step either increases its nucleophilicity via formation of an anion or generates in situ a reactive neutral intermediate after elimination of a leaving group, both of which allow coupling with the corresponding partner. In many instances, carbonate bases tend to be the best reagents. This outcome probably stems from their milder reactivity and greater functional group tolerance compared to stronger bases and will therefore be presented first.
Cyclic dienes (e.g., 2.1a) constitute very useful 4-atom synthons which have been coupled to three-atom components. For example, the reaction of 2.1a with α-halohydroxamates 2.1b, a precursor to aza-oxyallyl cations 2.1c, generates intriguing bicyclic compounds such as 2.1d (Scheme 2.1).[8] In this scenario, K2CO3 triggers an HBr elimination from 2.1b, giving rise to the reactive intermediate 2.1c. This work is just a recent example among many, exploiting a similar strategy, using inorganic bases to promote the (4+3)-annulations.[9] [10] However, in this review we will not cover the use of cyclic dienes as the reviews by Chiu, Mascareñas, and Harmata have discussed their use in (4+3)-annulations.

α-Halohydroxamates are an alternative motif that undergo formal annulations. Recently, Kim employed α-halohydroxamates 2.2b as aza-oxyallyl cation precursors with δ-hydroxy enones 2.2a in a Cs2CO3-mediated formal (4+3)-annulation for the synthesis of 1,4-oxazepan-3-one derivatives 2.2c (Scheme 2.2).[11] The use of a fluorinated solvent, trifluorotoluene, was crucial in order to attain high yields. The reaction tolerated both electron-rich and -deficient enones, with R1 corresponding to diversely substituted aryl and hetero(aryl) groups as well as simple esters (R1 = OMe and OBn). It was also possible to couple a series of different α-halohydroxamates (R2 = Me, Et, t-Bu and allyl). This (4+3)-formal annulation complemented the wider scope of a Na2CO3-mediated (4+2)-formal annulation within the same work, where δ-hydroxy enones 2.2a were replaced by their γ-hydroxy enone counterparts.
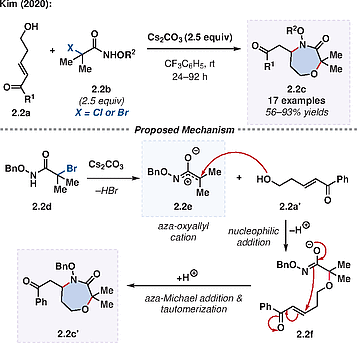
In the presence of Cs2CO3, elimination of HBr from 2.2d forms the key aza-oxyallyl cation 2.2e. The δ-hydroxy enone 2.2a′ then adds to 2.2e, providing the adduct 2.2f (Scheme 2.2). The latter undergoes an intramolecular aza-Michael addition followed by tautomerization to give the desired 1,4-oxazepan-3-one 2.2c′.
Another commonly used 4-atom synthon is the o-quinone methide (o-QM). This reactive intermediate can be generated in situ by a variety of approaches, one of which is activation by base. Stable o-QMs are generally limited to highly electron-rich examples. On the other hand, C,N-cyclic azomethine imines are stable 1,3-dipoles and have been extensively used in (3+2)[12] [13] [14] and (3+3)[15–17] annulations but infrequently in (4+3)-annulations. As such, an unprecedented and highly diastereoselective (4+3)-annulation between o-QM precursors, 2-(1-tosylalkyl)phenols 2.3a, and C,N-cyclic azomethine imines 2.3b for the construction of 1,3,4-oxadiazepanes 2.3c was developed in 2017 by Xu and Ren in the presence of the mild base Cs2CO3 (Scheme 2.3).[18]

Having either an electron-rich or an electron-deficient arene in the R1 position, such as in 2.3d and 2.3e, both resulted in ≥90% yield, whereas having a simple phenyl group (e.g., 2.3f) had a negative effect on the yield (Scheme 2.3). A methyl group in the same position was tolerated, e.g., 2.3g, but a substantial decrease in yield was observed when an ethyl group was used (e.g., 2.3h). Electron-donating and electron-withdrawing substituted phenols (R2) and imines (R3) were tolerated. A single example employing an N-tosyl-protected imine (R4) delivered the corresponding product 2.3i in a modest yield. The authors also incorporated the use of N,N′-cyclic azomethine imines 2.3j in the reaction (Scheme 2.3). In this case, the desired product cis-2.3k was formed in excellent yield but required a higher temperature for full conversion.
The high d.r. values were explained by differences in steric hindrance in two possible transition states: 2.4aTS and 2.4bTS (Scheme 2.4). The in situ formation of the o-QM by Cs2CO3-mediated elimination of TsH can either lead to the E- or Z-isomer. However, it is the E-o-QM that delivers the major diastereomer via the favored transition state 2.4aTS. This is due to greater steric hindrance between the p-Cl-phenyl moiety of Z-o-QM and the C,N-cyclic azomethine imine in the disfavored 2.4bTS, which hinders the annulation process. The favored 2.4aTS delivers the diastereomer cis-2.4 as the major product. Furthermore, the authors attempted to render the reaction enantioselective by employing a set of cinchona-derived bifunctional catalysts, albeit without any success.

In another instance, C,N-cyclic azomethine imines 2.5b were coupled with N-acetyl hydrazones 2.5a (Scheme 2.5).[19] The latter are precursors to 4-atom synthons called azoalkenes 2.5d, which are reactive species generated in situ via the base-mediated elimination of HX. In this case, K2CO3 was the optimal base for the reaction.

Both α-bromo- and α-chlorohydrazones (e.g., 2.5a, X = Br, Cl) gave the desired adduct, although most of the examples employed the former. Additionally, R1 was most often an aromatic ring, where both electron-donating groups (EDGs) and electron-withdrawing groups (EWGs) at various positions generated the corresponding products in good to excellent yields. A heteroaromatic (2-thiophenyl) and an aliphatic (t-Bu) hydrazone constituted additional substrates. Halogenated substrates (i.e., R2 = F or R3 = ClC6H4) were successfully employed. All but three examples used an N-benzoyl substituent at R3.
Although the reaction was not suited for N,N′-cyclized azomethine imines, this (4+3)-annulation was applicable to cyclic hydrazones 2.6a and 2.6b. When reacted with the standard azomethine imine 2.6c in the presence of K2CO3, the corresponding polycyclic products 2.6d and 2.6e were formed in modest yields (Scheme 2.6). Interestingly, a (3+3)-annulation took place when the N-benzoyl hydrazone 2.6f was reacted with 2.6c instead of the standard N-acetyl hydrazones 2.5a, delivering the triazine 2.6g (Scheme 2.6).

Most recently, and in analogy to the work by Chen and Xiao,[19] Hou and Gong[20] synthesized the sulfonylated analogues 2.7c of the tetrazepine derivates (Scheme 2.7). They generated an azoalkene 2.7d in situ using K2CO3 from 2.7a as well as the azomethine ylide 2.7e in situ from 2.7b using AgOTf as a Lewis acid catalyst in a one-step procedure.[20] The scope was found to be broad, and generally gave moderate to excellent yields for all substrates, regardless of substitution on 2.7a. The terminal alkyne was not tested in this reaction and only the phenyl substituent was used in that position. In a control study, the authors demonstrated that on using the preformed azomethine ylide 2.7e′ in the absence of AgOTf, the desired (4+3)-cycloadduct 2.7q could be generated in excellent yield.

Zhao also drew inspiration from Chen and Xiao’s work and use α-halogenohydrazones 2.8a and coupled them with nitrones 2.8b as the 1,3-dipoles. This delivered the 2,3,4,7-tetrahydro-1,2,4,5-oxatriazepine (4+3)-cycloadducts 2.8c with Na2CO3 as the mediating base (Scheme 2.8).[21]

The authors pointed out that the reaction of nitrones 2.8b resulted in a broader range of yields (45–99%) than those of the hydrazones 2.7b (64–90%). Most of the hydrazones 2.8a employed were brominated (X = Br) instead of chlorinated (X = Cl), as α-bromo hydrazones generally performed better. All but two hydrazones were N-acetylated (R2 = Me) and the remainder were limited to R2 = OMe as other substituents failed. Electron-poor arenes (e.g., 2.8d,e) gave significantly higher yields than electron-rich ones (e.g., 2.8f) (Scheme 2.8). For the nitrones 2.8b, electron-donating- and electron-withdrawing-substituted aryls at R3 were all well tolerated (e.g., 2.8g–i); heteroaryl, naphthyl and alkyl groups (e.g., 2.8j–l) were also compatible in the reaction. The majority of the examples had a –Bn group at R4, as the few examples with –Ph and –Me groups gave rise to lower yields. No reaction (NR) was observed when replacing the standard aldo-nitrone (H) by a keto-nitrone (Me) (e.g., 2.8m).
In the proposed reaction mechanism, the azoalkene 2.9b, generated in situ from 2.9a via Na2CO3-promoted HBr elimination, has two possible transitions states once it reacts with nitrone 2.9c (Scheme 2.9). DFT calculations allowed the authors to establish 2.9TS1 as the lower energy pathway due to the reduced steric repulsion between the benzyl of the nitrone and the phenyl group on the azoalkene. It was also determined that the formation of the desired seven-membered product 2.9d was kinetically favored.[21]

In 2018, both Li and Liu[22] and Shao[23] published similar base-mediated (4+3)-annulations between o-chloromethyl arylsulfonamides 2.10a and nitrones 2.10b for the synthesis of benzooxadiazepines 2.10c (Scheme 2.10). The former employed Na2CO3 whereas the latter employed NaHCO3. The o-chloromethyl arylsulfonamides are aza-ortho-quinone methide precursors (ao-QMs), which are generated in situ via a base-mediated HCl 1,4-elimination.

Coincidently, both reactions generated the standard product 2.10d in identical yields, with both authors’ substrate scopes tolerating a wide range of aryl substituents and a few alkyl substituents. However, only Shao’s work included substituents other than H at the R2 position, which resulted in the preferential formation of the cis over the trans diastereomer (e.g., 2.10e). Additionally, longer reaction times (i.e., 16 h) were needed when R2 = aryl, as conjugation within the intermediate structure of the azadiene deactivates it and therefore decreases the reaction rate.
Guo also employed o-chloromethyl arylsulfonamides 2.11a in an unexpected two-step (4+3)-annulation with arylcarbohydrazonoyl chlorides 2.11b for the construction of 2,3-dihydro-1H-benzo[e][1,2,4]triazepines 2.11d (Scheme 2.11).[24] The reaction proceeded via the formation of the intermediate 2.11c, which could be isolated and was initially the major product before flash column chromatography purification triggered its conversion into 2.11d. This led to silica gel being added as the second step of the reaction to completely consume 2.11c.

Various control experiments were carried out, since the obtained 2,3-dihydro-1H-benzo[e][1,2,4]triazepines did not clearly correspond to the expected products that would have arisen from a straightforward cyclization of intermediate 2.11c (Scheme 2.11). Interestingly, replacing silica gel with Lewis acids such as FeCl3 and MgSO4 also generated the same cycloadduct 2.11d′ (Scheme 2.11). However, protonic acids such as AcOH, PhCO2H and p-TsOH failed as substitutes. Based on these observations, the authors proposed that the silica gel increases the contact surface of intermediate 2.11c with the solvent, which helps the following transformation, while also activating 2.11c and 2.11e (vide infra).
Substitutions were tolerated in 2.11a except for when they were ortho relative to the NHTs group. For 2.11b, electron-rich aryl groups (Ar1) performed better than electron-deficient ones, and para-substituted aryl groups performed better rather than meta- or ortho-substituted ones, possibly due to steric hindrance. The same electronic trend was observed with regards to Ar2, although most examples only used a phenyl group at this position.
The authors proposed that this reaction occurred as a stepwise annulation process. First the electrophilic ao-QM 2.11e generated in situ from 2.11a′ via Na2CO3-mediated HCl elimination undergoes a conjugate addition with 2.11b′ to provide the key intermediate 2.11f. It is at this point that silica gel is added, which assists with the base-mediated deprotonation of 2.11f and causes the isomerization into the anion 2.11g. A subsequent intramolecular cyclization affords the spirocycle 2.11h, which rearranges and rearomatizes into the product 2.11d′.

In 2010, Hanson and co-workers developed a formal one-pot, cascade (4+3)-annulation for the synthesis of sultams 2.12c and 2.12e using Cs2CO3 (Scheme 2.12).[25] They employed epoxides 2.12a and sulfonamides 2.12b and 2.12d as ambiphilic synthons. These ambiphiles possess both an electrophilic and a nucleophilic site, which renders them ideal components for a complementary ambiphile pairing (CAP) cascade reaction.[25] After an initial epoxide opening, the resulting intermediate undergoes an intramolecular SNAr reaction in the case of α-fluorobenzenesulfonamides 2.12b or an intramolecular oxa-Michael addition in the case of vinylsulfonamides 2.12d. The reaction scope included a variety of allyl, benzyl, alkyl, and aryl substituents for R1 and R2. The R3 substituent was exclusively halogens and all products were generated in good yields.
It had previously been reported that epoxides efficiently undergo ring opening by sulfonamides when heated in 1,4-dioxane in the presence of catalytic amounts of tetra-n-butylammonium halides.[26] In other instances, BnEt3NCl had been used as a phase-transfer catalyst (PTC) under solid/liquid conditions in order to give high yields in short reaction times, with K2CO3 being employed as a base.[27] [28] This reactivity was ascribed to the potassium sulfonamide ion pair interactions, which are considerably smaller than the in situ formed quaternary ammonium pair.
When tert-butyldimethylsilyl (R)-(–)-glycidyl ether (2.13a) was reacted with N-allyl-2,6-difluorobenzenesulfonamide (2.13b), desilylation of intermediate 2.13c occurred, furnishing 2.13d with a free hydroxy group (Scheme 2.13). Mitsunobu reactions mediated by an oligomer coupling reagent, 2GOACC50, were performed to give rise to the corresponding sultam esters 2.13e in excellent yields.

Another (4+3)-annulation involving small-ring-opening was reported by Gong in 2015 (Scheme 2.14).[29] At the time, syntheses of the pyrrolo[1,2-a]azepin-9-amine skeleton in molecules had mostly been mediated by transition-metal catalysis with high catalyst loading.[30] [31] [32] [33] [34] [35] [36] [37] Gong et al. identified a way to access azepines 2.14c via a transition-metal-free (4+3)-annulation between ‘donor–acceptor’ reagents 2.14a and halocyclopropanes 2.14b under mild basic conditions using Cs2CO3. Compounds 2.14b are precursors to highly reactive cyclopropenes via a 1,2-elimination of HCl, and represent unique synthons with double nucleophilic and electrophilic centers via the cleavage of its C=C bond.

When the Ar3 moiety was substituted with EDGs at the para position, the corresponding azepines were obtained in good yields and excellent d.r. values (e.g., 2.14d,e). However, the opposite effect was observed with EWGs as both the yields and d.r. values dropped significantly (e.g., 2.14f,g).
EWGs on the Ar1 moiety tethered to the imine performed well in contrast to EDGs, which gave low yields and d.r. values (e.g. 2.14h-j). In this case, the reactivity of the imine is reduced towards nucleophilic attack by EDGs. This reduced reactivity logic applies to the single aliphatic imine example derived from n-butylamine, which resulted in an unidentifiable mixture of products. Bulky substituents seemed to constitute another important limitation: when Ar1 = biphenyl, the reaction gave rise to the lowest d.r. value (i.e., 7:1), and when R = tert-butyl on the ester moiety, a low yield of 58% was obtained.
Varying Ar2 led to similar observations with EDGs faring better than EWGs on the aryl moiety. However, strong EDGs (i.e., –Me) fared poorly. This pattern is likely due to the EWGs reducing the nucleophilicity of 2.14a, whereas strong EDGs decrease the acidity of 2.14a.

An indole-2-carboxaldehyde-derived donor–acceptor 2.15a was coupled with chlorocyclopropane 2.15b in an effort to expand the methodology (Scheme 2.15). After 0.6 hours, a formal (4+2)-annulation product 2.15c had formed. However, it was the desired azepino[1,2-a]indole 2.15d that constituted the major product after an hour of reaction time. This led the authors to postulate a mechanism where Cs2CO3 deprotonates 2.15a to give 2.15e, while the base triggers a 1,2-elimination of HCl from 2.15b (Scheme 2.15). The reactive cyclopropene 2.15f undergoes a conjugate addition to give rise to adduct 2.15g. An intramolecular Mannich reaction followed by protonation then occurs. The base deprotonates the resulting formal (4+2)-annulation intermediate 2.15h to generate the anion 2.15i, which undergoes a ring opening. Protonation of the seven-membered heterocycle 2.15j delivers the azepine 2.15k.
Though most examples of base-mediated (4+3)-annulations utilize carbonate bases, there are a select few which use hydride, oxide or amine bases.
The first example utilizing crotonate-derived sulfonium salts 2.16a as C3 synthons was developed by Yu, Wei and Meng in 2017 (Scheme 2.16, a).[38] The authors coupled 2.16a to thioaurones 2.16b to deliver 2,5-dihydrobenzo[4,5]thieno[3,2-b]oxepines 2.16c. Substrates with EWGs on the tethered Ar ring worked better than their electron-rich counterparts and no steric effects were observed. An isopropyl ester worked as well as a methyl ester group on 2.16a.

Subsequently, Peng and Li combined 2.16a with azadienes 2.16d in the presence of Cs2CO3 for the construction of tetrahydroazepino[2,3-b]indoles 2.16e (Scheme 2.16, a).[39] In some cases, a novel sulfide-catalyzed (4+3)-cyclization was also reported (Scheme 2.16, b). The products were generated in good to excellent yields with many different (hetero)aryl and aliphatic groups at R3. A single asymmetric example of the sulfide-catalyzed reaction was attempted with a camphor-derived sulfide, which gave rise to the enantioenriched cycloadduct 2.16e in 71% yield and 64% ee.
A control experiment was performed, where excess CD3OD was added to the reaction between 2.17a and 2.17b. The corresponding product D-2.17c incorporated 60% deuterium at both protons adjacent to the oxygen atom (Scheme 2.17). DFT calculations were also performed to provide insight into the reaction mechanism. The allylic ylide 2.17d is generated from deprotonation of 2.17a by NaH. Reacting the ylide with thioaurone 2.17b′ via a Michael addition forms the adduct 2.17e, which deprotonates the α-H to afford intermediates 2.17f. H/D exchange in the presence of CD3OD is supported by DFT calculations. A second proton exchange occurs to provide 2.17g before an intramolecular SN2 reaction affords the desired (4+3)-cycloadduct 2.17h.

In 2012, Mal reported the synthesis of 2-benzazepin-1-ones 2.18c from phthalides 2.18a and 2-azidoacrylates 2.18b using LiHMDS (Scheme 2.18).[40] In almost all cases, the cycloadducts 2.18c were oxidized in the presence of MnO2, furnishing the benzazepinediones 2.18d. The oxidized products 2.18d were easier to characterize by 1H NMR than compounds 2.18c.

The benzazepinones were generated from a variety of 2-azidoacrylates, which ranged from the parent methyl 2-azidoacrylate (2.18e), methyl 2-azidocrotonate (2.18f), an azido cyclic ketone (2.18g) and a series of 3-arylazidoacrylates (e.g., 2.18h). In the case of the phthalides, three out of four examples were characterized at the oxidized benzazepinedione form. Substrates containing a pyridine moiety (X = N) were successfully employed in the reaction (e.g., 2.18h).
The authors postulated that base-mediated deprotonation of phthalide 2.18a′ occurs at low temperature, affording the nucleophilic anion 2.18i, which attacks the 2-azidoacrylate 2.18j. The resulting adduct 2.18k can proceed via two potential pathways. Expulsion of N2 forms 2.18l, which cyclizes into the seven-membered 2.18m or undergoes direct attack at the lactone carbonyl to provide the bicycle 2.18n. The bicycle undergoes subsequent ring cleavage and the loss of N2 to access 2.18m. Tautomerization into 2.18o is followed by protonation, which yields the desired benzazepinone 2.18c′.
Qi and Jiang used t-BuOK in their (4+3)-annulation to furnish benzoxepines 2.19c from 2-fluorophenylacetylenes 2.19a and ketones 2.19b (Scheme 2.19).[41] The reaction tolerated various alkyl (hetero)aromatic and (cyclo)aliphatic ketones, though steric effects constituted an important limitation in the case of alkyl benzyl ketones. As for the 2-fluorophenylacetylenes, good to excellent yields were obtained whether the aromatic ring was electron-rich or poor. Furthermore, employing a chlorinated substrate in lieu of fluorinated 2.19a resulted in a complex mixture without 2.19c being formed.

To probe the mechanism of the reaction the authors stopped the reaction between standard 2.19a′ and 2.19d after an hour. They detected the β,γ-unsaturated ketone 2.19e′ in 48% yield along with the expected (4+3)-cycloadduct 2.19d′. After isolating 2.19e′ and submitting it to the standard reaction conditions, 2.19d′ was obtained in 78% yield. This indicated 2.19g constituted the key intermediate of the reaction and that C–C bond formation occurred before C–O bond formation.
These observations led the authors to postulate that base-promoted vinylation of 2.19f with 2.19a′ first occurs to give rise to key intermediate 2.19g. An ensuing deprotonation leads to (E)-enolate 2.19h, which isomerizes into its (Z)-counterpart. This allows an intramolecular SNAr process to happen, affording the desired benzoxepine 2.19i.
Another tert-butoxide-mediated (4+3)-annulation for the construction of benzoxepines 2.20c was reported by Wang who reacted 2-bromophenylacetonitriles 2.20a with ynones 2.20b in NMP (Scheme 2.20).[42] All but one of the R2 groups studied were (hetero)aryl and generally worked better when electron-rich. Electronic effects of the R1 and Ar groups had little impact on the yields. Changing the standard 2-bromo- to 2-iodo- and 2-chlorophenylacetonitrile 2.20a′ and reacting them with the standard ynone 2.20b′ resulted in a virtually identical yield of 2.20c′ for the iodinated substrate, whereas a much poorer yield was obtained using the chlorinated substrate (Scheme 2.20). Interestingly, changing the base and solvent to DBU and DMSO, respectively, changed the chemoselectivity and yielded an unexpected benzofuro[2,3-b]pyridine product via O-rearrangement.

The authors propose that t-BuOLi triggers the nucleophilic addition of 2.20a′ onto the alkyne, which forms intermediate 2.20d. A second deprotonation occurs, which affords enolate 2.20e. A subsequent intramolecular SNAr reaction delivers the desired benzoxepine 2.20c′.
Hua reported a hydroxide-promoted formal (4+3)-annulation between o-fluoro-N-alkyl-benzamides 2.21a and 2-propyn-1-ol 2.21 (Scheme 2.21).[43] The R1 substituents were exclusively (cyclo)alkyls and limited examples with substituents on the arene moiety (R2) were included, e.g., 3-Me, 4-MeO and 5-Cl. The solvent played an important role in the chemoselectivity. The use of DMSO led to the formation of 1,4-benzoxazepin-5(4H)-ones 2.21c, whereas MeCN generated (4+2)-adducts, i.e., 1,3-benzoxazin-4(4H)-ones 2.21f. As such, real-time monitoring by 1H NMR, carried out in DMSO-d 6 and CD3CN (not shown), was conducted to better understand the chemodivergent selectivities. Interestingly, the reaction between standard 2.21d and 2.21b delivered a mixture of the (4+3)- and (4+2)-adducts 2.21e and 2.21f during the first hours. The yield of the former increased over time as that of the latter decreased. Increasing the temperature from 30 to 50 °C gave 2.21f in <5%, whereas 2.21e was furnished in 82% yield. The authors suggested the base strength determined the chemoselectivity. KOH is completely soluble in DMSO, which creates a superbase and favors the rapid formation of 2.21e. In contrast, KOH is only slightly soluble in MeCN, creating a mildly alkaline solution, leading to the slower formation of 2.21f.

Based on these observations, it was postulated that a KOH-promoted SNAr reaction takes place between 2.21d and 2.21b to furnish the key intermediate 2.21g (Scheme 2.21). In DMSO, the strongly basic conditions promote the fast and favorable amide deprotonation. The resulting N-anion 2.21h undergoes an intramolecular nucleophilic addition onto the alkyne moiety via a 7-exo-dig cyclization. Subsequent protonation of the resulting allylic anion 2.21i leads to the (4+3)-adduct 2.21e. Alternatively, reversible allene formation involving 2.21j can occur in MeCN, and to some extent in DMSO, leading to the (4+2)-adduct 2.21f via a 6-exo-trig cyclization.
The final example comes from the Harmata group. They have had a long-standing interest in (4+3)-annulations involving allylic cations and their use in the synthesis of natural products.[44] [45] Building on the work of Katritzky in the late 1970s, thermal (4+3)-annulation reactions between N-alkylated oxidopyridinium ions and dienes have not been well developed, whereas N-arylated substrates perform exceptionally well in the reaction (Scheme 2.22, a).[46]

These scaffolds are substructures found in a number of alkaloids and facile methods to prepare them are of interest. To improve the reaction with N-alkyl substrates, Harmata utilized the ester-substituted pyridine 2.22d to enable the desired annulation reaction with both 2,3-dimethylbutadiene and 1,3-butadiene (Scheme 2.22, b).[47] DFT calculations predicted a concerted annulation. They also support an endo transition state TSA which is 0.7 kcal/mol lower in energy than the exo transition state TSB.
The authors explored 1-substituted and 1,2-disubstituted dienes to examine the regio- and stereoselectivity of the reaction (Scheme 2.23). In most cases, there was little endo/exo selectivity (2.23a–c). Regioselectivity was excellent in all cases with formation of a single regioisomer. Dienes containing carbocycles participated in the reaction and gave slightly better d.r. (e.g., 2.23d). When looking at 2,3-disubstituted dienes (e.g., 2.23e), regioselectivities were generally poor. One interesting example used the diene-tethered oxidopyridinium ion 2.23f to obtain a diastereomeric mixture of 2.23g and 2.23h.

The sole example where the endo product was strongly favored was with diene 2.24b (Scheme 2.24). In this case, the regioselectivity gradually degraded over the course of 48 hours. When each regioisomer was subjected to the reaction conditions, a similar mixture of 2.24c and 2.24d would arise indicating reversibility of the annulation (Scheme 2.24). Furthermore, subjecting either 2.24c or 2.24d to the reaction conditions with excess 2,3-dimethylbutadiene led to full conversion into the new cycloadduct 2.24e in both cases.

Base-mediated methods were broadly applicable but generally gave racemic products. One method to enable enantioselective transformations is to couple the base-mediated activation of one substrate with a chiral catalyst, such as an NHC, which we will cover in the next section.
3
Annulations Using N-Heterocyclic Carbenes
3.1N-Heterocyclic Carbenes (NHCs)
In general, NHC catalysts are reacted with aldehydes to create umpolung reactivity via the formation of a Breslow intermediate. The traditionally electrophilic aldehyde is turned into a nucleophilic conjugated enol that can attack an electrophilic partner. In the case of NHC-catalyzed (4+3)-annulations, chiral NHC catalysts ensure that the reactions can provide heterocycles in enantiomerically enriched form. The last step of the catalytic cycle usually involves a nucleophilic addition/elimination mechanism, where the positively charged NHC moiety linked to the restored carbonyl is ejected, thereby delivering the desired seven-membered ring and regenerating the active NHC.
In this past decade, the Ye group has made important contributions to this field. In 2013, they reported the first NHC-catalyzed (4+3)-annulation of β-monosubstituted enals 3.1.1a (R2 = H) with ortho-quinone methides (o-QMs) 3.1.1b to form the corresponding benzo-ε-lactones cis-3.1.1c (Scheme 3.1.1).[48] A few examples of the intermolecular reaction were reported, and even fewer enantioselective results. The authors stated that the typically unfavorable formation of a seven-membered ring is compensated by the driving force of re-aromatization and that this path outcompetes potential (3+2)- and (4+2)-annulations. Building on these findings, in 2015, Ye developed a (4+3)-annulation of enals 3.1.1a and aurones 3.1.1d with the same chiral triazolium catalyst 3.1.1aNHC to provide the corresponding benzofuran-fused ε-lactones cis-3.1.3e (Scheme 3.1.1).[49] In the following year, they were able to incorporate thiazolones 3.1.1f to yield the corresponding thiazole-fused ε-lactones 3.1.1g using near-identical conditions (Scheme 3.1.1, 2016).[50]

More recently, Ye incorporated both β-monosubstituted (R2 = H) and β,β-disubstituted enals (R2 = Me) 3.1.1a in a new (4+3)-annulation (Scheme 3.1.1, 2019).[51] Such a reaction was previously restricted to β-monosubstituted enals because of the steric encumbrance at the β-position. However, Ye previously reported methodologies that employed bifunctional NHCs with a free hydroxy group, e.g., catalyst 3.1.1bNHC, which proved crucial in generating the desired seven-membered benzofuroazepinones cis-3.1.1i using aurone-derived azadienes 3.1.1h as the enal annulation partner.
Ye’s (4+3)-annulation reactions via asymmetric NHC catalysis have greatly contributed to this field. The result is that a common set of general conditions can tolerate various substrate classes.
In the case of benzo-ε-lactones 3.1.1c, it is worth noting that a greater diastereoselectivities were observed for β-alkyl enals compared to β-aryl enals 3.1.1a. Substrates bearing a variety of substitution patterns on the tethered aryl moiety regardless of electronic effects, with the only exception being with a cinnamyl substituent (64% yield, 4:1 d.r.). Once again, β-alkyl enals were more successful than β-aryl enals, in terms of diastereoselectivity, with benzofuran-fused ε-lactones cis-3.1.1e being formed exclusively. Similar general observations were made about the reaction scope for the formation of the thiazole-fused ε-lactones cis-3.1.1g.
When aurone-derived azadienes 3.1.1h were reacted with β,β-disubstituted enals, >20:1 d.r. was typical except when R1 = R2 = Me. When R1 = an electron rich or poor aryl group, the products were generated in good yields and excellent ee values (e.g., 3.1.2a). Various substitution patterns on the tethered aryl moiety of the azadiene resulted in modest yields, but maintained excellent ee values (e.g., 3.1.2b). Interestingly, β-monosubstituted enals all resulted in lower d.r. (8:1 to 13:1 d.r., e.g., 3.1.2c). β,β-Dialkylenals gave the desired products in modest yields but maintained high ee values (e.g., 3.1.2d). Various aryl groups on the enal and the azadiene coupling partners were all tolerated.

The authors proposed that the NHC is bifunctional. The free hydroxy group proves to be crucial in directing the reactions towards forming seven-membered heterocycles, as opposed to forming spirocyclic cyclopentanones cis-3.1.3b and cis-3.1.3d via a competitive (3+2)-annulation. Control experiments revealed it was possible to transform ε-lactones cis-3.1.3a and cis-3.1.3c into the respective aforementioned spirocycles when employing 3.1.3aNHC and 3.1.3bNHC, which possess no free hydroxy group (Scheme 3.1.3). No reaction occurred when 3.1.1aNHC replaced 3.1.3aNHC, and neither was it possible to transform cis-3.1.3b into cis-3.1.3a. This outcome suggested the (4+3)-annulation is the kinetic pathway whereas the (3+2)-annulation is the thermodynamic.

A general reaction mechanism is outlined in Scheme 3.1.4, where the active catalyst 3.1.4NHC initially forms a vinyl Breslow intermediate 3.1.4b with enal 3.1.4a. Control of diastereo- and enantioselectivity is achieved via H-bonds between the carbonyl/imine of the 4-atom synthon and the hydroxy group of the NHC, which also increases the reactivity. Furthermore, an isopropyl group in the o-position on the arene of the triazolylidene moiety increases the bulk of the catalyst 3.1.4NHC. This interaction favors a subsequent Michael addition onto the o-QM-like substrate instead of a 1,2-addition to give enolate 3.1.4c. The latter undergoes Lewis basic O-acylation due to intramolecular H-bonding that enhances the Lewis acidity of the carbonyl. This interaction yields the kinetically favored seven-membered ring cis-3.1.4d and regenerates the active NHC catalyst. As such, thermodynamic Lewis basic C-acylation, which would yield spirocycles such as cis-3.1.3a and cis-3.1.3b, is thwarted.

By 2018, (4+3)-annulations for the construction of O- and N-heterocycles were well developed. Ye next explored sulfur heterocycles since these bioactive compounds are found in a great number of natural products. They reported an NHC-catalyzed (4+3)-annulation of bromoenals 3.1.5a with 1,2-benzenedithiol (3.1.5b) to produce benzo[1,5]dithiepin-2-ones 3.1.5c (Scheme 3.1.5).[52]

The reaction was successful with EDGs and EWGs at the para, meta and ortho positions of the β-aryl group of bromoenals 3.1.5a, where R1 = Ar and R2 = H (e.g., 3.1.5d) (Scheme 3.1.5). Aliphatic α-bromoenals gave the highest yields, where R1 = alkyl and R2 = H (e.g., 3.1.5e). The bromoenal where R1 = R2 = Me gave the corresponding thiolactone 3.1.5f in a high yield. The reactivity of a substituted 1,2-benzenedithiol 3.1.5b was not explored. Omission of 3.1.5aNHC led to the formation of dithiane 3.1.5g corresponding to the (4+2)-cyclization adduct. A control experiment revealed the latter could be transformed into thiolactone 3.1.5c′ in the presence of the NHC by ring expansion (Scheme 3.1.5). This result signified that the reactivity of the NHC differed from past observations as it did not immediately react with one of the substrates. The authors also explored the synthesis of six-membered heterocycles. Kinetic data for the (4+3)-annulation showed that 3.1.5g was generated in 90% yield in the first thirty minutes. It was only after extended time that ring-expansion took place to form 3.1.5c′ in 88% yield, with complete consumption of 3.1.5g after 9 hours.
The authors attempted to develop an enantioselective version, but 3.1.5bNHC generated 3.1.5c′ in only 34% ee (Scheme 3.1.5).
In the proposed mechanism, 1,2-benzenedithiol 3.1.6b first attacks enal 3.1.6a via a base-mediated Michael addition to give adduct 3.1.6c (Scheme 3.1.6). An intramolecular SN2 reaction occurs which generates the 1,4-dithiane 3.1.6d. Concurrently, 3.1.6NHC catalyzes the formation of the Breslow intermediate 3.1.6e. Cleavage of the C–S bond leads to acylazolium 3.1.6f, which undergoes intramolecular thio-lactonization to yield 3.1.6g and regenerate 3.1.6NHC.

Just prior to Ye’s work on 1,2-benzenedithiols, Lu and Du reported the first NHC-catalyzed asymmetric formal (4+3)-annulations of α,β-unsaturated acylazoliums derived from (Z)-2-bromoenals 3.1.7a with 2-aminobenzenethiols 3.1.7b and aryl 1,2-diamines 3.1.7d for the syntheses of 1,5-benzothiazepines 3.1.7c and 1,5-benzodiazepin-2-ones 3.1.7e (Scheme 3.1.7).[53] [54] While (4+3)-annulations had been extensively studied, the use of 1,4-bisnucleophiles with α,β-unsaturated acylazoliums remained unexplored. The aforementioned seven-membered heterocyclic moieties are found in many natural products and have prominent pharmacological and diverse biological activities. Additionally, unprotected aryl amines 3.1.7b and 3.1.7d are seldom used as nucleophiles in C(sp3)–N bond formation. These methodologies avoid an N-deprotection step and direct N-functionalization steps may be immediately applied. N-Protected bis- and mono-secondary benzene 1,2-diamines 3.1.8h and 3.1.8i failed to react (Scheme 3.1.8).

In the case of 1,5-benzothiazepines (e.g., 3.1.8a), it was observed that the steric and electronic properties of the substituents had limited impact on the reaction yields and ee values (Scheme 3.1.8). On the other hand, the reaction yields of 1,5-benzodiazepin-2-ones appeared to be influenced by the steric effect of the ortho substituents of the (Z)-2-bromoenal phenyl ring. Relatively large o-OMe 3.1.8b and o-Br groups resulted in considerably lower yields than the other substituted aryl ring examples; however, the ee remained high. Apart from these cases, monosubstituted (hetero)aryl, such as 3.1.8c, and naphthyl groups linked to the enal and symmetrical disubstituted aryl 1,2-diamines 3.1.8e were generally obtained in good yields and ee. Nonetheless, important limitations included the non-applicability of this reaction to alkyl-tethered enals as well as alkyl 1,2-diamines. The issue of regioselectivity was raised when unsymmetrical aryl 1,2-diamines were employed. In the case of meta-substituted diamines, a poor ratio was obtained (e.g., 3.1.8fa and 3.1.8fb) and the regioisomers were often inseparable by column chromatography. Nevertheless, the combined yields were high and the ee values remained excellent. On the other hand, ortho-substituted diamines gave products (e.g., 3.1.8d) with outstanding regioselectivity.

As a control experiment, (E)-3.1.9a was submitted to the standard reaction conditions with benzene-1,2-diamine (3.1.9b). The benzodiazepin-2-one 3.1.9c was only formed in 51% yield, as opposed to 96% yield when (Z)-3.1.9a was used (Scheme 3.1.9). This outcome demonstrated that the stereochemistry of 2-bromoenals strongly influenced the yield but not the enantioselectivity, which remained the same (Z: 97% ee, E: 97.5% ee). This outcome was also observed in the case of 1,5-benzothiazepines. Furthermore, β,β-disubstituted 2-bromoenal 3.1.9d was not a compatible substrate.

In the proposed catalytic cycle, the intermediate 3.1.10b, formed from active 3.1.10NHC and (Z)-3.1.10a, instigates the formation of 3.1.10c after tautomerization (Scheme 3.1.10). The latter is unstable and bromide elimination gives way to the in situ formed key reactive (E)-α,β-unsaturated acylazolium 3.1.10d. H-bonding is a key factor in generating enantioselectivity, as 3.1.10e attacks 3.1.10d via a 1,4-addition rather than through a 1,2-addition, which would lead to the same product via an alternative but racemic pathway. In the postulated transition state, 3.1.10e attacks from the Si face because the Re face is sterically obstructed by a large chiral group. Following tautomerization, chiral adduct 3.1.10f cyclizes into lactam 3.1.10g while regenerating the active 3.1.10NHC.

In a previous study of a thiourea-mediated (4+3)-annulation of in situ generated acylazoliums with 2-aminobenzenethiols, the authors surmised that the S-nucleophilic moiety of the latter first underwent a selective 1,4-addition onto the acylazoliums. Next, the N-nucleophilic moiety cyclized via an N-acylation step (see later on in Section 4 of this review).[55] This selectivity was taken advantage of in the synthesis of 1,5-benzodiazepin-2-ones.
The developments in (4+3)-annulations have led to increasingly complex structures. The enantioselective construction of spirocyclic indoles was one area of further investigation.
In the search for new reaction partners for (4+3)-annulations, Li et al. developed an enantioselective NHC-catalyzed reaction of ortho-hydroxyphenyl-substituted para-quinone methides (p-QMs) 3.1.11a with isatin-derived enals 3.1.11b for the synthesis of spirobenzoxopinones bearing an oxindole moiety 3.1.11c (Scheme 3.1.11).[56]

Electron-donating (e.g., 3.1.11d) as well as electron-withdrawing substitution (e.g., 3.1.11e) on the p-QM and enal aryl backbones gave good yields and high asymmetric induction (Scheme 3.1.11). The authors emphasize that there were no obvious electronic or steric effects. The less bulky diisopropyl-derived 3.1.11f also reacted well. However, no examples containing less bulky dialkylated p-QMs were included, probably because a competitive 1,4-addition side-reaction occurs instead of the desired 1,6-addition. Finally, oxindole 3.1.11g (lacking an N-protecting group) was also synthesized in good yield and excellent ee.
In analogy to Enders’ findings,[59] the double bond configuration of the enal 3.1.12a had no significant impact on the yield and asymmetric induction (Scheme 3.1.12). In another experiment, the presence of the TBS-protected hydroxy group in 3.1.12d prevented formation of the expected product 3.1.12f. Rather, the ene-ester 3.1.12e was the only observed product, suggesting that 3.1.12b does not directly react with 3.1.12a under the reaction conditions. Additionally, in an effort to demonstrate the synthetic potential of the reaction, both tert-butyl groups of 3.1.12c could be removed using AlCl3 in two steps.

It is proposed in Scheme 3.1.13 that Breslow intermediate 3.1.13b is formed from 3.1.13NHC and p-QM 3.1.13a. The azolium homoenolate intermediate then attacks 3.1.13c via a 1,6-addition to produce acylazolium 3.1.13d. The latter undergoes intramolecular cyclization to afford lactone 3.1.13e. No explanation was given as to how the chiral NHC induces the observed stereoselectivity.

The last example in this section describes the work of Chi from 2014. Though the synthesis of five- and six-membered dinitrogen-fused heterocycles via (3+2)- and (3+3)-annulations has been widely studied, this was not the case for seven-membered dinitrogen-fused heterocyclic derivatives 3.1.14c via (4+3)-annulations. Chi et al. therefore developed an NHC-catalyzed (4+3)-annulation of azomethine imines 3.1.14a and enals 3.1.14b with high diastereo- and enantioselectivity (Scheme 3.1.14).[57] This reaction is a rare example where the enal acts as the 4-atom rather than the 3-atom synthon. Additionally, the reactive sites of azomethine imine 3.1.14a are multiple bonds away from those of the NHC catalysts. Such an effective resolution was deemed impressive and thus a potentially useful strategy for challenging substrates.

The reaction worked effectively with azomethine imines 3.1.14a derived from various (hetero)aryl aldehydes and pyrazolidinones as well as with enals 3.1.14b having different (hetero)aryl substituents other than phenyl. One limitation was a significant drop in enantioselectivity when a vinyl substituent (cinnamyl) was tethered to the enal (84% ee).
The authors found that the enantiomers of racemic starting material 3.1.14a reacted differently in the presence of the chiral NHC-catalyst 3.1.14NHC. Therefore a kinetic resolution of azomethine imines could be carried out to obtain the enantiomerically enriched species (S)-3.1.14a (Scheme 3.1.14). Five examples were resolved with high selectivity factors (S = 60–339) by adding enal 3.1.14b′ in excess, all the while maintaining good yield and ee.

The authors postulate that formation of the Breslow intermediate 3.1.15b from active 3.1.15NHC and enal 3.1.15a is followed by oxidation by quinone 3.1.15[O] (Scheme 3.1.15). The resulting adduct 3.1.15c is deprotonated at its γ-carbon by K2CO3 to generate chiral vinyl enolate 3.1.15d, which acts as a 1,4-dipolarophile in the subsequent steps in a formal (4+3)-annulation. It first adds onto azomethine imine 3.1.15e to form 3.1.15f, which undergoes intramolecular cyclization to eject the seven-membered desired product while regenerating the active NHC catalyst. It is worth noting that it is the difference in reactivity between chiral 3.1.15d and the two enantiomers of 3.1.15e that allows for kinetic resolution.
3.2
N-Heterocyclic Carbenes and Base Dual-Activation
In many instances, NHC catalysis can easily be combined with base-activation of various substrates to promote (4+3)-annulations. In these situations, a dual-activation occurs, where the NHC reacts with the aldehyde substrate to form a Breslow intermediate while the base activates a secondary substrate. This allows the generation of two reactive intermediates in situ. This strategy has the advantage of using relatively stable starting materials to generate complex heterocyclic products. Additionally, because the formation of the NHC requires the addition of base, the two modes are highly compatible.
An excellent example of this concept comes from the work of Scheidt in 2013,[58] and related work by Ye on a near identical method.[48] Ye used preformed o-QMs, limiting the scope of the reaction to piperonal derivatives. The authors[58] stated that they had to perform extensive tuning of the reaction. Significant challenges had to be overcome such as NHC-Lewis base and NHC-o-QM compatibility, potential dimerization of the reactive transient o-QM and the obligation to have equilibrium populations of both the NHC–homoenolate and the o-QM. The authors used CsF/18-crown-6 to generate the o-QM from 3.2.1b which was then coupled with cinnamaldehyde derivative 3.2.1a, to afford the desired seven-membered ring lactones 3.2.1c (Scheme 3.2.1). A temperature of –18 °C and a reaction time of 12 hours were necessary to achieve optimal stereoselectivities. Interestingly, using the reactive acrolein 3.2.1d and the standard precursor 3.2.1b′ in the presence of a different NHC catalyst, 3.2.1bNHC, furnished the dihydrocoumarin 3.2.1e in good yield and excellent enantioselectivity.

Cinnamaldehydes bearing EDGs and EWGs participated in the reaction (3.2.1f–k), whereas only o-QM precursors with EDGs were effective. Additional substitution at the benzylic position of the o-QM precursors gave rise to vicinally substituted products 3.2.1l–n in good yields yet moderate diastereoselectivities.
In the proposed mechanism, the Breslow intermediate 3.2.1o is first generated from 3.2.1aNHC and cinnamaldehyde 3.2.1a′. Concurrently, the fluoride anion attacks the TBS group on 3.2.1b′, forming the corresponding o-QM via a desilylation/elimination process. A C–C bond-forming event ensues via a 1,4-addition from the less-hindered side of the NHC–homoenolate onto 3.2.1b′-o-QM. After tautomerization of the resulting adduct 3.2.1p, an intramolecular O-acylation releases the 7-membered lactone 3.2.1c′ and regenerates 3.2.1NHC.
In 2016, Enders et al. focused attention on the NHC-catalyzed direct asymmetric synthesis of seven-membered spiro-N-heterocycles. Two classes of products were at the heart of their research: spirobenzazepinones 3.2.2c generated atroposelectively from isatin-derived enals 3.2.2a and N-(ortho-chloromethyl)aryl amides 3.2.2b (Scheme 3.2.2), as well as spiro-1,2-diazepinones 3.2.4c generated from the same category of enals and α-halo hydrazones 3.2.4b (see Scheme 3.2.4).[59]

Precursors 3.2.2b and 3.2.4b are converted in situ into aza-o-QMs and azoalkenes, respectively, via Cs2CO3-mediated 1,2-elimination of HCl. Both reactions tolerated a wide range of electronically and sterically substituted substrates to provide the corresponding products in good ee values.
The authors postulated a plausible catalytic cycle for the synthesis of spirobenzazepinones. Enal 3.2.2a′ and 3.2.2NHC′ react together to give the Breslow intermediate 3.2.2d. Its mesomeric form 3.2.2e reacts with in situ generated aza-o-quinone 3.2.2b′ via a 1,4-addition to build a quaternary stereocenter in acylazolium 3.2.2f. Intramolecular cyclization yields lactam 3.2.2c′ and regenerates the active catalyst 3.2.2NHC′.
The removal of the N-tosyl group or reduction of the carbonyl group on the azepinone ring of spirobenzazepinone 3.2.3a to form 3.2.3b and 3.2.3c, respectively, eliminated atropisomerism. The authors also measured a coalescence and interconversion of 3.2.3a atropisomers above 90 °C in DMSO-d 6 (Scheme 3.2.3).

The mechanism for the synthesis of spiro-1,2-diazepinones is similar to that of spirobenzazepinones. The double bond configuration of 3.2.4a′ was shown to have no significant impact on the yield or ee of spiro-1,2-diazepinones 3.2.4c′ (Scheme 3.2.4). Compound 3.2.4a′ was also coupled to cyclic hydrazones to give polycyclic spiro-1,2-diazepinones 3.2.4d, 3.2.4e and 3.2.4f, as well as to an α-chloro-oxime to give spiro-1,2-oxazepinone 3.2.4g.

In 2014, Glorius was able ‘to selectively generate different products from identical substrates, utilizing catalyst rather than substrate control’. Indeed, by simply modifying the NHC catalysts, enantioenriched 1,2-diazepines 3.2.5c and pyrazoles 3.2.5d were formed with good chemo- and regioselectivity from enal 3.2.5a and α-chloro hydrazone 3.2.5b (Scheme 3.2.5).[60] During the optimization, it was surmised that the electronic and steric differences of the catalysts 3.2.5aNHC and 3.2.5bNHC were responsible for the change in annulation mechanism. Using N-Boc hydrazones was also essential for the selectivity and good yields as other N-protecting groups were not as efficient.

Enals 3.2.5a bearing para-substituted electron-donating and electron-withdrawing aryl groups (R1) generated modest to good yields (40–70%). The best outcome was obtained with 3.2.5a′ and a p-Br group resulted in a low yield of 40%. A β-2-furyl and two alkyl enals were also well tolerated. Various substituents at para and ortho positions of aryl (R2) hydrazones 3.2.5b were compatible, as were 2-naphthyl and tert-butyl hydrazones. Cyclic hydrazones such as 3.2.5e gave lower ee (85%, 87% ee) compared to linear ones (≥93% ee). Employing a less bulky NHC catalyst, where the N-Mes moiety is replaced by N-p-Tol, delivered higher asymmetries (93%, 97% ee), but with poor yields (21%, 25%). The authors derivatized 3.2.5a′ by removing the N-Boc group with TFA and, separately, performed a direct hydrogenation to reduce the imine functionality.
The homoenolate 3.2.6b is generated from 3.2.6NHC and enal 3.2.6a. Meanwhile, the azoalkene is formed in situ by means of elimination of HCl from hydrazone 3.2.6c. A 1,4-addition occurs where 3.2.6b attacks the electrophilic 3.2.6c-azoalkene with the help of a H-bonding interaction between the two species. The resulting azolium 3.2.6d undergoes an intramolecular N-acylation to provide the desired 1,2-diazepine 3.2.6e and the regenerated NHC catalyst (Scheme 3.2.6).

Hui coupled 2-bromoenals 3.2.7a with 1,4-bisnucleophilic N-Ts hydrazones 3.2.7b for the construction of chiral tetrahydro-1H-1,2-diazepines 3.2.7c (Scheme 3.2.7).[61]

The majority of differentially substituted (hetero)aromatic groups gave rise to good yields and excellent ee values, although electron-rich 2-bromoenals and electron-deficient N-Ts hydrazones tended to be the more efficient substrates; all the reported examples resulted in >20:1 d.r. Alkyl-substituted 2-bromoenals were not compatible with the asymmetric version of this reaction.
The authors were also able to generate the standard tetrahydro-1H-1,2-diazepine 3.2.8c via a different methodology with the help of the commonly used oxidant 3.2.8[O] in NHC-catalyzed (4+3)-annulations (Scheme 3.2.8). Coupling N-Ts hydrazone 3.2.8b with the in situ formed acylazolium intermediate derived from cinnamaldehyde 3.2.8a and the NHC catalyst was possible.

The α,β-unsaturated acylammonium species 3.2.9b created from 3.2.9NHC and enal 3.2.9a forms H-bonds to the conjugate base of N-Ts hydrazone 3.2.9c (Scheme 3.2.9). This interaction helps the subsequent stereoselective Michael addition to occur with the C-nucleophilic hydrazone moiety, which forms adduct 3.2.9d. A proton transfer leading to another acylazolium 3.2.9e leads to a final lactamization step, which releases the desired tetrahydro-1H-1,2-diazepine 3.2.9f and regenerates active 3.2.9NHC.

Fu and Huang developed a (4+3)-annulation reaction mediated by chiral 3.2.10NHC, where oxotryptamines 3.2.10a and enals 3.2.10b were coupled together in the presence of the stoichiometric oxidant 3.2.10[O] to give rise to enantioenriched spiro-ε-lactam oxindoles 3.2.10c (Scheme 3.2.10).[62] This was the first time flexible oxotryptamines were employed to construct seven-membered heterocycles.

The d.r. was strongly influenced by the size of R1. Bulkier groups increasingly led to improved selectivity (i-Pr > Et > Bn > Me). Various substituents on the oxotryptamine ring (R2) and different arenes at the β-position of the enals were tolerated, furnishing the corresponding products in generally good yields and asymmetries. One example, using a β-methyl enal, successfully generated the corresponding product, albeit in poor yield.
In the proposed mechanism, formation of a Breslow intermediate between chiral 3.2.11NHC and enal 3.2.11a was followed by oxidation by [O] to afford acylazolium 3.2.11b (Scheme 3.2.11). Simultaneously, deprotonation of oxotryptamine 3.2.11c under basic conditions gives rise to the corresponding enolate. The latter attacks 3.2.11b via a Michael addition to form intermediate 3.2.11d. Cyclization delivers spiro-ε-lactam oxindole 3.2.11e and the regenerated catalyst.

The same authors were also able to develop a one-pot process leading to enantiopure hexahydropyrroloindoles 3.2.12b, which are privileged structural motifs found in various pharmaceuticals and natural products with promising biological activities (Scheme 3.2.12). The oxindole 3.2.12a underwent ring opening with methanol before reductive cyclization furnished the desired products in good to excellent yields.

In summary, the NHC-catalyzed (4+3)-annulations described use enals as the 3-atom synthons, except for a single instance. The 4-atom partners are varied yet share an (aza)-o-QM or (azo)diene-like structure. In some cases, the choice of the NHC was crucial in order to form the desired seven-membered rings, as different ring sizes or spirocycles could be generated.
4
Phosphines
In the past few decades, phosphines have emerged as one of the most widely used classes of organic catalysts. They are most renowned as nucleophilic reagents with many unsaturated compounds, including alkenes, allenes, alkynes, and Morita–Baylis–Hillman (MBH) alcohol derivatives. The zwitterionic compounds can act as versatile nucleophiles and engage in various kinds of annulations.[63] In particular, allenoates and MBH derivatives are most often used in this class of (4+3)-annulations.[64] The reactions in this section will be categorized based on those two substrate classes and one interesting example will be described using phosphazenes as active species.
Guo and Kwon explored the phosphine-catalyzed (3+2)-annulation of N,N-cyclic azomethine imines 4.1a with allenoates 4.1b. They also determined that allenoates with a second carboxylate group at the β′-position undergo (3+3)- and (4+3)-annulations (Scheme 4.1).[65] They were able to show that bulky alkyl groups on the phosphine made the (4+3)-annulation more favorable, providing a 3:1 mixture of the two diastereomeric tetrahydropyrazolodiazepinones 4.1e and 4.1f in a combined yield of 63%, alongside (3+2)-product 4.1c and (3+3)-product 4.1d.

They proposed that the phosphine adds to the β-position of 4.1b, forming the zwitterionic phosphonium enoate 4.1g. Steric hindrance from the butyl chains on the phosphine hinders the (3+2)-annulation. Less bulky phosphines were proposed to avoid these interactions. The second carboxylate group at the β′-position enhances the acidity of the β′-protons, which enables an intramolecular proton transfer, affording structure 4.1h. Nucleophilic addition forms amide 4.1i. This intermediate can either engage in a 6-endo cyclization, ultimately yielding the (3+3)-annulation product 4.1d, or undergoes a tautomerization that leads to structure 4.1j. A 7-endo cyclization provides phosphonium ylide 4.1k, while subsequent proton transfer and elimination of the phosphine catalyst yields the two diastereomeric (4+3)-annulation products 4.1e and 4.1f.
In 2012, Guo and Kwon examined phosphine-catalyzed annulations between C,N-cyclic azomethine imines 4.2a and allenoates 4.2b (Scheme 4.2).[66] When R2 of 4.2b was an aromatic ring, the results were consistent with the findings of their previous work: (3+2)-Annulation was predominantly observed when PMe3 was used as the catalyst whereas the use of PBu3 led to (4+3)-annulation. However, when R2 of 4.2b was an alkyl group, the substrates underwent (3+2)-annulation regardless of the catalyst used.

The (4+3)-annulation delivered a single diastereomer of the hexahydrodiazepinoisoquinoline derivatives 4.2d in moderate to excellent yields, along with small amounts of the (3+2)-product 4.2c. EWGs on the aromatic substituent R2 of 4.2b generally led to higher yields. Various substituents on the aryl ring of 4.2a delivered good yields, but led to the formation of considerable amounts of the (3+2)-product.
The proposed mechanism is similar to the previous example and starts with nucleophilic addition of the phosphine catalyst to 4.2b, forming phosphonium enoate 4.2e, which then attacks 4.2a to afford amide 4.2f. After tautomerization into intermediate 4.2g, the amide performs a 7-endo cyclization leading to structure 4.2h, from which the phosphine catalyst is regenerated to yield the product 4.2d.
It was not until 2016 that Guo reported the enantioselective version of the reaction using a chiral phosphine catalyst. Using C,N-cyclic azomethine imines 4.2i with allenoates 4.2b, catalyzed by chiral Kwon phosphine P1,[67] they synthesized a broad range of diversely substituted tetrahydrodiazepinoquinazoline derivatives 4.2j in good to excellent yields and with good d.r. and ee values. The electronic effects of the substituents on the R2 group of 4.2b appeared to have no effect on the yield or selectivity of the reaction, but substituents at the ortho-position lowered the diastereoselectivity. When R2 = Me, the product was formed in only 70% ee. When R2 = H, the yield dropped to 70%, while the enantioselectivity was retained. When substituents were introduced at position 6 of the quinazoline moiety of 4.2i, the reaction was successful (4.2k,l), whereas a bromine at position 7 led to a dramatic decrease in diastereoselectivity (4.2m). In general, steric aspects seem to have a larger impact than electronic effects. Examples of possible further functionalization of the products 4.2j were also reported. For example, selective reductions could be performed as well as an oxidative ring opening of the quinazoline ring.
These routes rely on the phosphine catalyst activating a neutral four-atom synthon to attack a 1,3-dipolar three-atom synthon. Zheng and Lu described the opposite approach, activating a three-atom synthon to react with a 1,3-diene as a four-atom synthon. In 2009, following up on the work of Ishar et al.,[68] they reported the phosphine-catalyzed (4+3)-annulation reaction of ethyl allenoate 4.3b with methyl coumalate (4.3a), forming substituted cycloheptatriene 4.3d along with two regioisomers of the (3+2)-cycloadduct (Scheme 4.3). They performed the same reaction with allylic carbonates 4.3c as a three-atom synthon, yielding bicyclo[3.2.2]nonadienes 4.3e.[69] This result was one of the early examples using methyl coumalate in a (4+3)-annulation, as well as one of the first using phosphine catalysis.

While they did not succeed in optimizing the conditions for the allenoate, so as to suppress the formation of the (3+2)-cycloadduct, they were able to achieve this selectivity for the reaction with allylic carbonates 4.3c. These allylic carbonates are easily accessible from acrylates, acrylonitriles or vinyl ketones and aldehydes through the Morita–Baylis–Hillman (MBH) reaction.[70] Zheng and Lu introduced different aryl- and alkyl-substituents on the MBH carbonates, resulting in moderate to good yields of highly substituted compounds 4.3e. The electronic effects of the substituents on the aryl moiety seemed to have no effect on the yield and replacement of the ethyl ester group by a t-butyl ester or a methyl ketone was successfully employed. Replacing the methyl ester group on the coumalate by N,N-diisopropylamide or a nitrile group lowered the yield to 43% and 65%, respectively.
The proposed mechanism begins with the addition of PPh3 to 4.3c, eliminating the Boc-protected allylic hydroxy group to form tert-butoxide, carbon dioxide and allylic phosphonium derivative 4.3f. Deprotonation of 4.3f by tert-butoxide leads to the zwitterionic structure 4.3g, which adds to 4.3a, giving adduct 4.3h. A 7-endo cyclization affords intermediate 4.3i, from which the final product 4.3e is formed by elimination of the phosphine catalyst. A similar intermediate 4.3j, formed in the reaction of the allenoate with 4.3a, can undergo decarboxylation before the phosphine catalyst is eliminated to give 4.3d.
In 2017, Huang followed up Zheng and Lu’s work and reported a sequential domino (4+3)-annulation/allylic alkylation of N-tosyl azadienes 4.4a with MBH carbonates 4.4b, yielding multifunctionalized benzofuran-fused seven-membered heterocycle derivatives 4.4c in generally excellent yields (Scheme 4.4). Screening of different additives revealed a significant increase in yield when benzoic acid was added in catalytic quantities, though no information was disclosed on the mechanistic origins of this improvement. Remarkably, a second equivalent of the MBH carbonate is attached to the newly built seven-membered ring, with both reactions catalyzed by trianisylphosphine. The reaction tolerated diversely substituted aryl groups on the exocyclic double bond of the azadiene.[71] However, an EWG at position 4 of the core benzene ring or EDGs at the para-position of the annulated aryl group of the azadiene lowered the yields significantly. Different esters or a nitrile group on the MBH derivative were generally tolerated in good to excellent yields. Furthermore, the authors successfully demonstrated the scalability of the reaction without a decrease in yield.

The first steps of their proposed mechanism are identical to the mechanism presented by Zhang and Lu (see Scheme 4.3). The phosphine catalyst adds to 4.4b, eliminating the allylic Boc-group and forming allylic phosphonium derivative 4.4d (Scheme 4.4). Deprotonation by the tert-butoxide leads to zwitterionic structure 4.4e, which then adds to 4.4a in a nucleophilic attack to give 4.4f. The azepine ring is then formed via 7-endo cyclization, eliminating the phosphine catalyst. The benzofuran annulated dihydroazepine 4.4g is deprotonated by tert-butoxide present in the reaction mixture to afford carbanion 4.4h with a negative charge on the carbon atom bearing the carboxylate group. The carbanion attacks a second equivalent of 4.4d, eliminating the phosphine catalyst in an SN2′ reaction to yield the final product 4.4c.
In a subsequent report in 2019, Huang et al. presented a system that performed a phosphine-dependent (4+3)-annulation. When trianisylphosphine was used, the substrates underwent a sequential domino (4+3)-annulation/allylic alkylation.[72] The reaction yields fluoroalkyl-substituted, fully-dehydrogenated benzazepines 4.5d,e in good yields, using trifluoroacetimidoyl chlorides 4.5a and MBH carbonates 4.5b in combination with triphenylphosphine or trianisylphosphine as catalysts (Scheme 4.5, a). Both catalysts promote the (4+3)-annulation, leading to the common intermediate 4.5c, but only trianisylphosphine is able to initiate the allylic alkylation to form allyl-substituted benzazepine 4.5d. With triphenylphosphine, only base-catalyzed isomerization to benzazepine 4.5e takes place. Both reactions tolerated fully fluorinated methyl and ethyl groups on 4.5a and different ester groups on 4.5b. Substrates with EWGs at the para-position to the imidoyl group provided the highest yields for both reactions. Interestingly, naphthyl groups on the imidoyl moiety were only successfully employed in the annulation with triphenylphosphine. A bromine substituent at the ortho-position blocked the (4+3)-reaction completely. The authors succeeded in scaling-up the reaction to gram scale, as well as selectively reducing different functional groups of the products.

The authors performed mechanistic studies in support of the proposed catalyst dependence of the alkylation reaction. Intermediate 4.5c′ can be trapped by adding only one equivalent of cesium carbonate and performing the reaction at room temperature (Scheme 4.5, b). Heating 4.5c′ with base, but no phosphine catalyst gave 4.5e′ in almost quantitative yield. When starting material 4.5b was present, the yield dropped, but no alkylation product was observed. However, heating 4.5c′ with 4.5b and trianisylphosphine led to 4.5d′ in excellent yield, regardless of the presence of cesium carbonate.
In analogy to the previous mechanisms, the cycle begins with the nucleophilic addition of the catalyst to MBH carbonate 4.6a, forming the zwitterionic phosphonium enoate 4.6b. Addition to the imidoyl group of acetimidoyl chloride 4.6c forms amide 4.6d (Scheme 4.6). Chloride is eliminated from this intermediate, giving ketimine 4.6e. Deprotonation provides zwitterionic structure 4.6f, possessing a delocalized negative charge that can engage in a 7-endo dearomative cyclization to afford structure 4.6g. Elimination of the phosphine catalyst gives intermediate 4.6h, which can rearomatize by tautomerization to yield isolatable intermediate 4.6i. As the mechanistic study suggested, base-mediated tautomerization leads to final product 4.6j, while deprotonation affords structure 4.6k, from which addition to activated MBH carbonate 4.6l gives alkylated product 4.6m.

Notably, there have been non-catalytic examples of phosphine-mediated (4+3)-annulations which relate to this topic. In 2015, Chen et al. reported that brominated MBH derivatives 4.7a (Scheme 4.7, a), prepared from isatins 4.7f as electrophiles in the MBH reaction (Scheme 4.7, b), can be used in a (4+3)-annulation with in situ generated aza-o-quinone methides 4.7e to prepare aza-spirocycloheptane oxindole scaffolds 4.7c in moderate to good yields.[73] However, a stoichiometric amount of the phosphine and ten equivalents of base were needed to generate phosphonium enoates 4.7d quantitatively. Bromo-substituted MBH derivatives formed from aryl aldehydes with methyl acrylate were not compatible with the reaction under the specified conditions. EDGs on 4.7a led to better yields than EWGs. Introducing substituents on the N-(o-chloromethyl)aryl amide 4.7b led to moderate yields, regardless of the electronic effects of the newly introduced substituent. A benzyl group was also successfully incorporated as a protecting group on the nitrogen atom of 4.7a. The ethoxycarbonyl protecting group of 4.7b could be replaced by a Boc-group, but a tosyl group was not compatible with the reaction. The (4+3)-cycloadducts could then be subjected to deprotection and selective reduction. The use of chiral catalysts to make the reaction enantioselective was not successful.

In 2019, Budynina et al. followed a completely different approach in phosphine-mediated (4+3)-annulations, reporting a new domino self-catalytic Michael/aza-Wittig reaction between phosphazenomalonates 4.8b and acrolein towards hexahydroazepines (Scheme 4.8, a).[74] The phosphazenomalonates are generated in situ from the corresponding azidomalonates 4.8a and triphenylphosphine via a Staudinger reaction. The phosphazene moiety first acts as an intramolecular catalyst for the Michael addition of the malonate moiety to acrolein, before being used as a reactive functional group in an aza-Wittig reaction with the aldehyde, closing the seven-membered ring. The tetrahydroazepines 4.8d were characterized in crude form as they proved to be unstable to column chromatography materials such as SiO2 and Al2O3. As a result, the crude products were reduced prior to purification, affording diverse hexahydroazepines 4.8e in moderate to good overall yields, independent from electronic effects of functional groups on the (hetero)aryl substituents (R) of 4.8a.
The application of phosphazenes in self-catalytic systems is a novel approach. To investigate the details of the catalytic system, the authors conducted a detailed mechanistic study. To learn whether the Michael addition or aza-Wittig reaction happens first, they ran a competition experiment between 4.8a′, the corresponding azido-monoester 4.8f and only one equivalent of acrolein, with the monoester being unable to perform the Michael addition under the selected conditions (Scheme 4.8, b). After two hours, 1H NMR showed comparable amounts of the phosphazene species 4.8b′ and 4.8g, but the ratio between annulation product 4.8d′ and aza-Wittig product 4.8h was 20:1. This led the authors to suggest that the Michael addition is the rate-determining step and that it occurs before an intramolecular aza-Wittig reaction. Furthermore, experiments in the absence of triphenylphosphine showed no reaction, while the combination of triphenylphosphine and a dummy substrate without an azide group led to polymerization of acrolein. The otherwise unreactive 4.8g was able to catalyze the Michael addition when added in catalytic amounts to the experiment with the dummy substrate, although it was gradually consumed by the aza-Wittig reaction. This leads to the conclusion that the phosphazene moiety is indeed the active species in the catalytic cycle.

An experiment with the isolated Michael adduct 4.8a′, acrolein, and triphenylphosphine led to intense tarring, which the authors explained by oligomerization due to the drastic increase in the concentration of phosphazene units during the fast Staudinger step, occurring in the presence of active aldehyde moieties. This outcome supported that the order of the steps is crucial. When acrolein was exchanged for methyl vinyl ketone, the Michael addition was slowed down, and the aza-Wittig reaction did not proceed under standard conditions. Upon heating, the phosphazene 4.8i underwent a 1,5-aza-Wittig reaction with the ester, forming dihydropyrrole derivative 4.8j (Scheme 4.8, c). A DFT study supported 1,5-cyclization with the ester group being kinetically favored over the 1,7-cyclization with the keto group. Notably, the transition state of the rate-determining step of the 1,7-aza-Wittig reaction between the phosphazene and formyl group in 4.8c is ΔH = 8 kcal/mol lower than the transition state for the 1,5-aza-Wittig reaction with the ester group, making the 1,7-cyclization more favorable.
In conclusion, phosphine catalysis represents a very useful tool to activate specific classes of substrates, including allenoates or Morita–Baylis–Hillman carbonates, to participate in (4+3)-annulations with a broad variety of unsaturated or dipolar synthons through similar mechanisms. However, a recent example of a domino Michael/aza-Wittig reaction using phosphazenes as intramolecular catalysts of both reactants showed that there are still more pathways to be discovered that involve phosphine catalysis. Another advantage of phosphine catalysis proved to be the fact that there is an enormous library of chiral phosphines available, enabling the exploration of stereoselective versions of the annulations.
5
Acids
In this section, we will survey the literature regarding Lewis acid and Brønsted acid catalyzed and mediated reactions. The Lewis acids in question can contain transition metals, but often can be used interchangeably with other Lewis acidic metal salts and at times, even with Brønsted acids to enable the same transformation. The Brønsted acids section will include methods using chiral phosphoric acids, which enable an enantioselective transformation, as well as the use of achiral acids, which render racemic products.
5.1Lewis Acids
Lewis acids have long been known for their capacity to activate by complexation to Lewis basic sites.[75] [76] The reactions described in this section will be sorted by both the type of metal used (In, Ga, Fe, Zn, Y, Yb, Sc, Ti) and the structural moieties participating in the reaction. Although a separation between main group and transition-metal Lewis acids may seem evident, multiple examples will illustrate that strict categorization is not straightforward and that some reactions can work with various kinds of Lewis acids. In the end of the section, three examples are presented, using Lewis acids in stoichiometric amounts or in tandem with Brønsted acids and applying the concept in the total synthesis of three natural products.
In 2017, Selander examined various InX3-catalyzed transformations of alkynyl enones 5.1.1a (Scheme 5.1.1) with in situ generated enamines.[77] When InBr3 or InCl3 were used and the enamine was formed from cyclopentanone 5.1.1b and morpholine, they were able to obtain bicyclo[3.2.1]alkenones 5.1.1c in very good yields. It was necessary to add 4 Å MS to suppress the formation of hydroamination product 5.1.1d. Replacing cyclopentanone with cyclohexanone and morpholine with pyrrolidine led to good yields of the analogous bicyclo[3.3.1]alkenones. Other Lewis acids such as In(OTf)3, AgOTf and ZnCl2 were also catalysts for this reaction, although the yields were lower. Notably, the reaction also took place when no catalyst or only catalytic amounts of a Brønsted acid were added, albeit resulting in lower yields and accompanied by the formation of more side product.

When examining the scope of this reaction, the authors found that the electronic properties of the R1 substituents on an aryl group of 5.1.1a had no significant effect on the yield of the reaction, while alkyl groups at the R1 position led to moderate yields. Substitution of the aryl group by a methyl group at the R2 position decreased the yield significantly. R3 = methyl, phenyl and methoxy groups were all tolerated. When positions R2 and R3 were connected by an ethylene or propylene linker, tricyclic structures were formed under the standard conditions. Attempts to perform the reaction enantioselectively using chiral amines instead of morpholine were not successful.
According to the proposed mechanism, the Lewis acid coordinates to the π-system of the alkyne and a lone pair of the oxygen atom in the carbonyl group of 5.1.1a′, activating the β-position of the Michael system for the nucleophilic attack of enamine 5.1.1e. The addition product 5.1.1f rearranges into 1,2-allenyl ketone 5.1.1g through Lewis acid alkyne activation. Product 5.1.1c′ is formed by an intramolecular enamine addition at the allene carbon, followed by hydrolysis. Although the amine is in theory regenerated, experiments with catalytic amounts remained unsuccessful. The authors believe that the amine is not fully liberated under the reaction conditions, explaining why it is used in excess.
Wu et al. reported a catalytic regio- and diastereoselective three-component (4+3)-annulation in 2012, using either InBr3 or GaBr3 as the Lewis acid.[78] Remarkably, the reaction proved to furnish cyclohepta(b)indoles 5.1.2d in excellent yields, starting from readily available indoles 5.1.2a, carbonyls 5.1.2b and conjugated dienes 5.1.2c, at ambient temperature and without the need for an inert atmosphere (Scheme 5.1.2). The corresponding triflates showed even higher catalytic activity, but the formation of byproducts or decomposition was observed. Some Brønsted acids proved to be suitable catalysts as well, but their use led to lower yields and more byproducts.

Both unprotected and N-alkylated indoles 5.1.2a participated in the reaction. EDGs on the indole, as well as on 5.1.2b, accelerate the reaction rate. EWGs were also tolerated. Non-cyclic dienes 5.1.2c gave significantly lower yields. The diastereomeric ratio of the products varied between no selectivity and >10:1, with electron-poor aldehydes generally leading to lower diastereoselectivity. The use of an unsymmetrical diene led to a single regioisomer.
The authors proposed that 5.1.2a adds to the carbonyl group of 5.1.2b, forming the alcohol 5.1.2e. Elimination of the hydroxy group leads to indolyl cation 5.1.2f that can undergo annulation with 5.1.2c. DFT studies led the authors to the conclusion that the actual annulation is most likely a two-step process. In the first step, a C–C bond is formed between the indolyl cation and a terminal diene carbon atom, giving intermediate 5.1.2g, which can then undergo intramolecular ring closure to yield product 5.1.2d.
Ghorai et al. demonstrated the temperature-modulated diastereoselective dimerization of 2-vinylindoles 5.1.3a to form tetrahydrocarbazoles 5.1.3b and tetrahydrocycloheptadiindoles 5.1.3c (Scheme 5.1.3).[79] When a solution of 5.1.3a in DCE was exposed to catalytic amounts of various FeIII salts at 30 °C, 5.1.3b was produced through a (4+2)-annulation between two molecules of the 2-vinylindole. Fe(OTf)3 proved to possess the highest catalytic activity, giving very good yields and diastereoselectivity. When the reaction temperature was raised to 50 °C, they observed the formation of 5.1.3c through a (4+3)-annulation. At 80 °C, compounds 5.1.3c were formed exclusively in excellent yields and as single diastereomers. Methyl and halogen substituents on the aryl ring were tolerated, as well as methyl and ethyl esters on the olefinic moiety. The reaction proceeded with an unprotected indole, although the presence of a methyl or allyl N-protecting group led to higher yields. A substrate lacking an ester group on the vinylic moiety gave the (4+2)-annulation product regardless of the temperature.

The authors propose that the FeIII-based Lewis acid coordinates to the indolyl nitrogen of one molecule of 5.1.3a, reversing the polarity of the 2-vinyl group in the generated intermediate 5.1.3d. Another molecule of 5.1.3a can attack the vinyl group with its C3 center, affording intermediate 5.1.3e. A 6-endo-cyclization and subsequent rearomatization leads to (4+2)-annulation product 5.1.3b, whereas an alternative 7-exo-cyclization is followed by rearomatization leading to product 5.1.3c.
In 2014, Liang et al. built a furan ring and a seven-membered ring in one step, using ZnCl2 as a catalyst, by combining Knoevenagel adducts 5.1.4a and tert-butyldimethylsilyl-protected butadien-2-ol derivates 5.1.4b (Scheme 5.1.4).[80] Cyclohepta(b)furans 5.1.4c were isolated in moderate to good yields with ZnCl2, while other ZnII and PtII salts gave lower yields.
Steric effects significantly impacted the reaction, for example o-substituted aryl rings led to only traces of the corresponding product 5.1.4c, whereas substrates with substituents at meta and para positions were successfully employed. EWGs on the aryl ring also lowered the yields, compared to EDGs. Substrates with alkyl substituents on the alkyne failed to react. Aryl groups next to the carbonyl groups in 5.1.4a did not affect the reaction, whereas removal of the methyl substituent on 5.1.4b decreased the yield. Furan proved to be an unsuitable diene for this reaction.

The authors postulated two possible mechanisms, having furyl zinc carbene 5.1.4e as a focal intermediate. This carbene 5.1.4e is generated by coordination of the ZnII catalyst to the alkyne moiety of 5.1.4a, activating it for a 5-exo-cyclization to give a furyl zinc carbene 5.1.4d. Theoretically, the carbene could now either (i) form a cyclopropane intermediate, which could then undergo a (3,3)-sigmatropic/Cope rearrangement[7] [81] [82] to close the seven-membered ring, or (ii) undergo a (4+3)-annulation with the diene via transition state 5.1.4e. To determine whether the cyclopropanation/Cope pathway was viable, the cyclopropane intermediate was synthesized via a different route and was subjected to the reaction conditions. No product was detected, suggesting the latter mechanism is more likely. Considering that ZnCl2 can catalyze Knoevenagel reactions, the authors attempted to prepare 5.1.4c′ in a one-pot reaction directly from pentane-2,4-dione (5.1.4h), alkynal 5.1.4i and 5.1.4b. They obtained the desired product with a yield of 15% under the standard conditions, though no further attempts were taken to optimize the reaction.
Two years later, Liang investigated the reaction of 2-azidomethyl-indoles 5.1.5a with trisubstituted propargyl alcohols 5.1.5b in the presence of Zn(OTf)2 (Scheme 5.1.5).[83] They found that various metal triflates were effective catalysts for this reaction, with Al(OTf)3, Yb(OTf)3, Bi(OTf)3 and In(OTf)3 giving lower yields and Y(OTf)3 giving the best yields. The conditions were then optimized using Y(OTf)3, and the tricyclic indole azepine scaffold 5.1.5c was obtained in good yields. Numerous groups were tolerated on all three aryl substituents of 5.1.5b, regardless of their electronic properties. Substrates with R2 = alkyl on the alkyne moiety gave the corresponding indole azepines in good yields. Secondary alcohols and alkyl-substituted tertiary alcohols were not compatible with this reaction. Various halogens and methoxy substituents were tolerated as R2.

According to their proposed mechanism,[84] [85] the Y(OTf)3 catalyst coordinates to 5.1.5b, forming intermediate 5.1.5d. Protonation enables elimination of water through attack by the C3 carbon of 5.1.5a on the alkyne, giving cationic allene derivate 5.1.5e. Rearomatization through deprotonation and nucleophilic attack of the negatively charged nitrogen atom of the azide group on the allene leads to intermediate 5.1.5f. Elimination of N2 and a proton affords structure 5.1.5g, from which the Lewis acid is regenerated through protonation, yielding product 5.1.5c.
In 2011, Pagenkopf et al. used Lewis acidic rare-earth element triflates as catalysts for the (4+3)-annulation of nitrones 5.1.6a with donor–acceptor cyclobutanes 5.1.6b, relying on the ring strain released by opening the cyclobutane ring in 5.1.6b (Scheme 5.1.6).[86] Using 10 mol% of Yb(OTf)3, they obtained oxazepines 5.1.6c in excellent yields and as the pure cis-diastereomers within an hour. However, when the catalyst loading was decreased and the reaction was stopped after ten minutes, two different diastereomers were found. Subjecting the mixture of the two diastereomers to Yb(OTf)3 led to quantitative isomerization of the trans- to the cis-diastereomer. Varying the temperature, catalyst loading and equivalent ratios of the educts led to different ratios of the two diastereomers, but longer reaction times eventually led to the pure cis-diastereomer under all conditions. Notably, the reaction did occur in the absence of a Lewis acid, as long as 4 Å MS were added. Both diastereomers were formed in a cis/trans ratio of 1.3:1.0 and in this case, isomerization was not observed.

The reaction time needed to give a single diastereomer was strongly dependent on the electronic properties of the R1 substituent on 5.1.6a. Electron-rich nitrones led to shorter reaction times. Fortunately, the yield was not dramatically affected directly by electronic effects, although longer reaction times led to higher amounts of decomposition and background reactions. In addition to aryl groups, various heteroaromatics, a cinnamyl group and a naphthyl group were successfully incorporated in 5.1.6a. Notably, an N-benzyl nitrone with R1 = H was a suitable substrate as well. Various R2 aryl substituents and a benzyl group were also tolerated. On 5.1.6b, a fused six-membered cyclic ether worked as well as a non-cyclic ethyl ether, although this resulted in a mixture of diastereomers.
In 2017, Wang et al. used the same catalyst in their studies on an intramolecular (4+3)-annulation. They reported the cyclization of a (3)dendralene-tethered cyclopropane-1,1-diester 5.1.7a, yielding vinyl-substituted (5.3.0)decane derivates 5.1.7b that could be further derivatized through subsequent (4+n)-annulations (Scheme 5.1.7).[87] The vinyl group of the dendralene moiety not participating in the first annulation not only enables subsequent derivatizations of the product, but it also stabilizes the carbocation in intermediate 5.1.7d through delocalization and thus facilitates the reaction. The products 5.1.7b were obtained in good to excellent yields as a single diastereomer, accompanied by the side product 5.1.7c, formed through (3+2)-annulation. Ratios varied between 4.5:1 and >20:1. This observation led the authors to propose an SN2 mechanism for the (4+3)-annulation. They suggest a nucleophilic attack of the internal C–C double bond of the dendralene moiety on the cyclopropane, resulting in ring opening through a Walden inversion. Nucleophilic attack of the carbon atom bearing the geminal esters on the central carbon of the pentadienyl cation leads to a 7-endo-cyclization to form the product.

Diene 5.1.7b readily underwent (hetero)-Diels–Alder reactions with various dienophiles, including alkyne diesters, maleimides, 1,4-benzoquinone and nitrosobenzene. An attempt to combine the (4+3)-annulation and the Diels–Alder reaction into a one-pot sequence was also successful. Additionally, some more exotic derivatization reactions were possible. The reaction of 5.1.7b′ with Fe(CO)9 gave adduct 5.1.7e, which was then able to react with dibromo isobutyrone in a second (4+3)-annulation to give tetracyclic scaffold 5.1.7f. The use of enantiopure starting material 5.1.7a′′ led to the selective formation of 5.1.7b′′, which was then able to undergo a stereospecific (6+4)-annulation with tropone to afford the pentacyclic structure 5.1.7g as a single enantiomer.
A ring-opening (4+3)-annulation, using cyclopropane derivatives and a rare earth element triflate as a Lewis acid catalyst, was reported by Werz et al. in 2017.[88] They subjected 1,3,5-triaryl-1,3,5-triazinanes 5.1.8a and cyclopropane-1,1-diesters 5.1.8b to various types of Lewis acids, of which Sc(OTf)3 gave the highest yield of 1,3-diazepane derivatives 5.1.8c (Scheme 5.1.8, a). When enantiopure chiral cyclopropane derivatives were used, only one enantiomer of the product was formed via a stereospecific reaction with inversion of the stereocenter.

The authors proposed a mechanism that started with coordination of the Lewis acid to the geminal diester moiety of 5.1.8b, giving 5.1.8d, which underwent nucleophilic attack by 5.1.8a through an SN2 mechanism. The resulting linear zwitterionic iminium intermediate 5.1.8e is formed through two simultaneous ring openings while inverting the stereocenter of the former cyclopropane. Direct ring closure of this intermediate would afford a nine-membered ring and is therefore unfavorable. The elimination of one equivalent of formaldimine shortens the chain by two atoms and allows the resulting intermediate 5.1.8f to undergo a 7-endo-cyclization, affording the final product 5.1.8c.
A competition experiment with differentially substituted triazinanes did not yield any crossover product, which led the authors to the conclusion that the triazinane precursors do not decompose into three molecules of formaldimine under the reaction conditions.
The reaction proved to tolerate a vinyl group and a broad range of (hetero)aryl substituents at position R1 on 5.1.8b, as well as various substituted aryl groups on the nitrogen atoms of 5.1.8a. When subjected to HCl in methanol, the heterocycles 5.1.8c were hydrolyzed to afford linear 1,4-diamines 5.1.8g in good to excellent yields (Scheme 5.1.8, b). Combining the annulation and hydrolysis into a one-pot sequence proved feasible.
Recently, Werz used similar conditions with Sc(OTf)3 to enable a (4+3)-annulation between cyclopropane-1,1-diesters 5.1.9a and thiochalcones 5.1.9b to afford tetrahydrothiepines 5.1.9c diastereoselectively (Scheme 5.1.9).[89] Using enantiopure cyclopropanes, the reaction was shown to be stereospecific, completely transferring the stereochemical information during the transformation, while inverting the stereocenter of the substrate. Without heating the reaction, tetrahydrothiophene derivates 5.1.9d formed as side products via a (3+2)-pathway. When ether-based solvents or a titanium-based Lewis acid were used, the (3+2)-cycloadducts were the sole products. A broad range of functionalized (hetero)aryl substituents with various electronic properties were tolerated on both the cyclopropane and the thiochalcone. On the cyclopropane, a succinimide and a vinyl group proved to be compatible, whereas a phthalimide group showed only low conversion. According to the proposed mechanism, activated 5.1.9e undergoes an SN2 reaction with the nucleophilic sulfur atom of 5.1.9b, in analogy to the previous example. The resulting zwitterionic intermediate 5.1.9f possesses a delocalized positive charge and can undergo ring closure to form either the five-membered (3+2)-adduct 5.1.9d or the desired (4+3)-adduct 5.1.9c. Subjecting isolated 5.1.9d to the reaction conditions showed that the formation of the five-membered ring was reversible, as it isomerized quantitatively into the thermodynamically favored seven-membered ring 5.1.9c.

In 2015, Morgan et al. reported a BF3·Et2O-mediated Nazarov cyclization of allenyl vinyl ketones 5.1.10a, intercepted by a (4+3)-annulation with dienes 5.1.10b, to build a bicyclo[4.2.1]nonene ring system 5.1.10c (Scheme 5.1.10, a).[90] Bicyclo[2.2.1]heptenes 5.1.10d were also formed in varying quantities through a (3+2)-annulation. When butadiene and 2,3-dimethylbutadiene were used as dienes, only the (4+3)-cycloadducts were formed. Introducing substituents on the ends of the diene led to increased amounts of the (3+2)-product, and dienes bearing two substituents on one end follow the (3+2)-pathway. This result can be explained by steric hindrance in intermediate 5.1.10f. According to the proposed mechanism, the reaction proceeds through a Lewis acid mediated Nazarov cyclization[91] [92] of 5.1.10a, forming intermediate 5.1.10e. Instead of deprotonation, this intermediate suffers nucleophilic attack by 5.1.10b to give zwitterionic intermediate 5.1.10f, which can then undergo a ring closure and elimination of the Lewis acid to yield the product 5.1.10c.

The final ring closure proved to be reversible, as the (3+2)-annulation product 5.1.10d was able to isomerize into thermodynamically more stable products when subjected to the reaction conditions for an extended time (Scheme 5.1.10, b). Depending on the substituents, either 5.1.10c, a bicyclo[5.1.2.1]nonene scaffold 5.1.10h or the ring-opening product 5.1.10g was formed. However, considerable amounts of the substrate decomposed under the strongly acidic conditions, making the isomerization inefficient.
There are limited examples of total syntheses using a Lewis acid catalyzed (4+3)-annulation approach, though the transformation has the potential to generate complex fused rings in a single step. As one of the first examples, Winne et al. reported the reaction of dienes 5.1.11b with furfuryl cations 5.1.11d, generated from furfuryl alcohols 5.1.11a and TiCl4 (Scheme 5.1.11).[93] Although this reaction uses stoichiometric amounts of Lewis acid, it is included due to the link with subsequent mechanistic studies and total syntheses described below. The resulting dihydrocyclohepta(b)furans 5.1.11c were obtained in moderate to excellent yields. A substituent at the 5-position of 5.1.11a proved to be essential, as substrates lacking a substituent led to immediate polymerization. Primary furfuryl alcohols gave very low yields, whereas secondary alcohols gave very good yields and tertiary alcohols gave almost quantitative yields. This trend can be explained by a better stabilization of reactive intermediates. Secondary furfuryl alcohols showed increasing exo-selectivity for the double bond with increasing size of the substituent on the alcohol. Various linear, cyclic and cyclo-fused dienes participated. DFT analysis of a stepwise vs a concerted mechanism led the authors to the conclusion that the stepwise mechanism is consistently favored. They proposed that a Lewis acid mediated elimination of the hydroxy group leads to the stabilized furfuryl cation 5.1.11d. The cation is then attacked by 5.1.11b to form allylic cation 5.1.11e. 7-endo-cyclization gives intermediate 5.1.11f and deprotonation affords product 5.1.11c.

Winne’s group applied this reaction in the synthesis of the two cyclohepta(b)furan-containing natural products frondosin B [(+)-5.1.12a] (Scheme 5.1.12) and epi-liphagal [(+)-5.1.13a] (Scheme 5.1.13).[94] They were isolated from marine sponges and multiple groups have contributed total synthesis studies towards these compounds.[95] The synthesis of liphagal is of particular interest, as it selectively inhibits one isoform of the phosphatidylinositide 3-kinase (PI3K) enzyme family, which is involved in cellular signaling processes.[96] The substrates 5.1.12b,c and 5.1.13c,d can be synthesized in a few steps from commercially available compounds.

When subjecting substrates 5.1.12b and 5.1.12c to stoichiometric amounts of TiCl4, a complex reaction mixture was obtained, from which small amounts of racemic O-methyl frondosin B 5.1.12d and two minor diastereomers 5.1.12e were isolated. All three were intermediates in previous total syntheses of frondosin B and could be converted into the natural product through demethylation and tautomerization with BBr3. The formal total synthesis of frondosin B was therefore successful, although the yield and selectivity were not satisfactory. The use of FeCl3·6H2O instead of TiCl4 led to a different tautomer of O-methyl frondosin B (5.1.12f) in a much cleaner reaction, but the yield was still very low. Interestingly, a bis-adduct structure 5.1.12g was also isolated, which is formed through a Friedel–Crafts-type reaction. When trifluoroacetic acid was used instead of the Lewis acids, a better yield of 5.1.12g was obtained, which could then be successfully deprotected and isomerized into the racemic final product, frondosin B (5.1.12a), in one step with BBr3. Including the synthesis of the substrates 5.1.12b,c, the racemic total synthesis was completed in four steps.
For the total synthesis of epi-liphagal [(+)-5.1.13a], compounds 5.1.13c and 5.1.13d were subjected to TiCl4, which led to trace amounts of the desired product 5.1.13b. The predominant product was 5.1.13e, which is formed through irreversible proton elimination before the slower cyclization step can occur. This proposal was supported through DFT calculations. The use of CF3CO2H instead of TiCl4 as the cation-generating reagent made this deprotonation reversible, resulting in very good yields of the desired intermediate (+)-5.1.13b, with only small amounts of 5.1.13e. The reaction also proved to be highly stereoselective and gave mainly the natural configuration of liphagal. Surprisingly, no Friedel–Crafts product was observed. The authors explained this with the decreased electrophilicity of the bis(methoxy)benzofurfuryl cation, compared to the mono(methoxy)benzofurfuryl cation. Although the total synthesis looked very promising until this step, the last step proved to be more challenging than expected. Efforts to reduce the isolated double bond in the cycloheptane ring to that found in liphagal failed, as all attempts led to the hydrogen being introduced on the opposite side of the tetracycle. However, the introduction of the aldehyde group was successful, ultimately leading to an unnatural diastereomer of liphagal.

A recent report by Rawal employs an atypical two-step strategy to enable a ‘(4+3)-annulation’ towards the synthesis of the core scaffold of (–)-ambiguine P (Scheme 5.1.14).[97] Although model studies using simpler dienes worked well with TMSOTf in CH2Cl2, the desired diene only formed the first C–C bond. Further reaction in the presence of NaAuCl4·2H2O in i-PrOH afforded the desired cycloadduct. This example highlights the observations of Winne and shows limitations to the existing methodology.

Lewis acid catalyzed (4+3)-annulations with various Lewis acids have been developed in recent decades, and the scope is as diverse as the pool of appropriate synthons. This methodology allows the synthesis of highly substituted, multi-fused ring systems with a broad range of heteroatom substitutions in specific patterns, but it also reveals the main drawback: most reactions are highly substrate-specific and not generalizable. One of the reactions proved to be useful for the total synthesis of two natural products. Although most of the reactions are diastereoselective and some are stereospecific, there have been no examples of enantioselectivity.
5.2
Brønsted Acids
In many instances, (4+3)-annulations using Brønsted acids with an alcohol generate the carbocation along with a molecule of H2O. This type of acid-mediated activation of furfuryl alcohols was studied by Winne (Scheme 5.2.1).[98] For the synthesis of the rameswaralide scaffold, the reaction was performed using TFA in H2O to yield the desired multicyclic fused system (Scheme 5.2.1, a).[99] More recently, work towards the daucane sesquiterpenoid skeleton used TFA in CH2Cl2 to generate the desired cycloadduct in excellent yield and as a single diastereomer (Scheme 5.2.1, b).[100] Furthermore, heterocycles other than furan could be incorporated using this method. In work towards the synthesis of the giberellane and kauranoid scaffolds, Ga(OTf)3 (20 mol%) or TFA (2 equiv) could be used in CH2Cl2 to afford the desired cycloadducts, but in poor diastereoselectivities (Scheme 5.2.1, c).[101] [102] These examples illustrate that both solvent and acid play subtle roles in the reaction.

It is evident that although numerous strategies exist to synthesize the 5,7,6-fused-ring scaffold, interest in preparing liphagal and frondosin B remains strong. Xue and Li used a similar strategy employing the benzofuran 5.2.2a and diene 5.2.2b in the presence of (±)-camphorsulfonic acid (CSA) (Scheme 5.2.2).[94] [103] The use of a Brønsted acid did not improve the diastereoselectivity of the reaction and a 1:1 mixture of the anti/syn (4+3)-cycloadducts was observed. This cycloadduct could then be used to carry out the formal synthesis of (±)-epi-liphagal and (±)-frondosin B.[104]

In 2016, Shi demonstrated the use of 3-indolylmethanols 5.2.3a in a (4+3)-annulation reaction with α,β-unsaturated ketones 5.2.3b using HBr in CH3CN to form spirocyclic cycloadducts 5.2.3c (Scheme 5.2.3).[105] Within 1 minute in the presence of the acid, water is lost and the adduct is formed in 86% yield. This species could follow two possible pathways to obtain the desired cycloadducts. The first pathway proposes a [1,3]-C migration of the nucleophilic carbon center to the 2-position of the indole to form carbocation 5.2.3e. Following this step, a deprotonation will generate 5.2.3f. Michael addition will then afford the desired cycloadduct 5.2.3c. A second pathway is the retro-Michael addition of 5.2.3d′ in the presence of HBr. This would generate a stable carbocation species, which could undergo a 1,2-insertion with 5.2.3b. Upon generation of 5.2.3f, a Michael addition would form 5.2.3c.

Substitutions on either the oxindole or indole ring on the indole substrate 5.2.3a were possible (Scheme 5.2.4). Notably, halogen substitutions at the ortho-position of the oxindole ring or the 6-position of the indole ring afforded good yields of the cycloadducts (71–85%). In contrast, all other substitutions on the other positions of the rings gave moderate yields (49–65%).

In 2018, the Lautens group disclosed a [4+3]-annulation between an in situ generated o-QM 5.2.5d and an isomünchnone 5.2.5e to yield the syn oxa-bridged oxazocine cores 5.2.5c, generating N2 and H2O as the sole by-products (Scheme 5.2.5).[106] Using only a catalytic amount of CSA, it was possible to generate both reactive intermediates in one step, eliminating the need for the rhodium catalysts generally employed for isomünchnone formation.

Both 6- and 7-membered diazoimide substrates participated in the reaction, with the larger ring size affording slightly better yields (5.2.5f–g,r,s). Furthermore, an acyclic diazoimide reacted, albeit in lower yield (5.2.5q). Electronic effects on the aryl substituents did not follow a pattern as both OMe and Cl fared worse compared to a Me substituent (5.2.5k–o). Interestingly, a OMe substituent on the aryl ring of the o-QM gave a high yield (5.2.5h).
The use of chiral phosphoric acids (CPAs) can enable an enantioselective (4+3)-annulation. For example, Jiao and Shi recently reported the (4+3)-annulation between 2-indolylmethanols and o-QMs to give cyclohepta[b]indoles.[107] The authors proposed that the o-QM was formed initially in solution and reacted with the indolylmethanol to first form the C–C bond. Following the loss of a second molecule of H2O from the 2-indolylmethanol, the C–O bond would form to generate the desired (4+3)-cycloadduct (Scheme 5.2.6).

To this end, the cycloadducts could be generated with moderate to excellent ee values and yields. Interestingly, slight modification of the reaction conditions allowed for the rarely used tertiary benzylic alcohols to be suitable as substrates for the annulation, albeit affording lower yields and lower ee values of the desired cycloadducts.
Another method for the enantioselective synthesis of cyclohepta[b]indoles was reported by Masson in 2018 (Scheme 5.2.7). Masson presented the catalytic asymmetric (4+3)-annulation using a H8-CPA, setting three stereocenters in a single step.[108] Similarly, a 3-indolylmethanol 5.2.7a was used in combination with a dienecarbamate 5.2.7b to generate the cycloadduct 5.2.7c.

Following the loss of H2O from 5.2.7a, an ion pair was presumed to be formed between the CPA catalyst and the newly formed indolium species. The dienecarbamate 5.2.7b can undergo hydrogen bonding with the phosphoryl oxygen to ensure a highly organized transition state. The authors proposed that attack on the Re face of the indole and Si face of the diene were favored. This facial selectivity would create a boat-like conformation for the ring-closing step to afford the all-cis product as the major isomer. In the absence of an R5 group, there would be free rotation around the newly formed C–C bond, leading to lower diastereoselectivity (5.2.7d,e). However, high ee values were retained throughout. Due to the mild conditions, substrates bearing terminal alkenes (5.2.7g) and boronic esters (5.2.7j) could be reacted, albeit in lower yields.
In 2017, Shi reported the highly diastereoselective (4+3)-annulation between o-QMs or aza-o-QMs and N,N′-cyclic azomethine ylides using 10 mol% of a phosphoric acid catalyst in PhMe at 80 °C in the presence of 5 Å MS (Scheme 5.2.8).[109] Numerous Brønsted acids were tested including AcOH, TFA, TsOH, TfOH, MsOH, HBr and HCl, but none gave the desired cycloadduct. The use of CPAs under various conditions led to only 28% ee.

The primary focus was the use of o-QMs as substrates, which afforded the cis products in moderate to excellent yields and generally excellent diastereoselectivities. Interestingly, when aza-o-QMs were used as the substrates, the trans product was strongly favored (>95:5 d.r.). The authors propose the reason for this change in stereoselectivity was due to the amine group. Orientation of the bulky amine group in the trans conformation, compared to the Ar group of the N,N′-cyclic azomethine ylide led to lowered steric congestion for the annulation.

Recently, Shi also reported a (4+3)-annulation between N,N′-cyclic azomethine imines 5.2.9b and isatoic anhydrides 5.2.9a involving the extrusion of CO2 by using a phosphoric acid catalyst (10 mol%) (Scheme 5.2.9).[110] The authors proposed that this reaction proceeds via a stepwise pathway. Following the loss of CO2, a zwitterionic acylium species 5.2.9d forms and reacts with the N,N′-cyclic azomethine imine 5.2.9b. The resulting acylium species 5.2.9e undergoes an intramolecular 6-exo-dig cyclization to generate spirocyclic intermediate 5.2.9f. This rigid structure had a high tendency to undergo a subsequent ring opening and 7-endo-trig cyclization resulting in the observed cycloadduct 5.2.9c.
The authors demonstrated that although the cycloadduct could be formed through a three-component reaction between 5.2.9a′, 5.2.9h and benzaldehyde (5.2.9i), the yield was poor. Further experiments showed that a minimal amount of 5.2.9b is formed from the combination of 5.2.9h and 5.2.9i under the standard reaction conditions.
In summary, the use of Brønsted acids has been shown to be compatible with a variety of substrates, although each set of conditions is highly specific to the desired transformation.[110] The reactions often employ a substrate which can liberate a molecule of H2O or N2 to generate a reactive intermediate. Hence, this mode of activation is often used with substrates containing benzyl alcohols, indolylmethanols or diazo functional groups. Stoichiometric amounts of Brønsted acids such as HBr, TFA or CSA are often used and generate racemic products. When a single enantiomer of CSA was used, it did not impart any chiral information into the (4+3)-cycloadducts. In comparison, excellent enantioselectivities could be achieved with CPAs in both polar and apolar solvents under mild reaction conditions.
6
Boronic Acid Catalysis and Photocatalysis
In 2015, Zheng reported a method to enable (4+3)-annulations between indole/benzofuran derivatives 6.1a and 1,3-dienes 6.1b. 3,5-Bis(trifluoromethyl)phenylboronic acid was used as a catalyst to synthesize valuable cyclohepta[b]benzofurans, cyclohepta[b]benzothiophenes and cyclohepta[b]indoles 6.1c (Scheme 6.1).[111] Syntheses of this scaffold have been previously accomplished by Wu,[78] Winne,[93] Hsung[112] and Li,[113] but this report was one of the first to use boronic acid catalysis. The authors noted that they tested various Lewis and Brønsted acids as well as protic acids with similar pK a values as the boronic acid chosen, but none enabled the desired (4+3)-annulation.

The use of phenylboronic acid did not afford the desired product, likely due to weak acidity. However, electron-deficient boronic acids were tested and led to the formation of products 6.1c under mild reaction conditions. The scope was limited, but illustrated that O-, N- and S-heterocycles participated in the reaction as well as both cyclic and acyclic dienes, affording the products in moderate to excellent yields (e.g., 6.1d–h). In the case of 6.1i, which was synthesized from the 3-substitued indole, when the reaction was run with 1 equivalent of the boronic acid, the yield increased to 89% with identical diastereoselectivity. The authors proposed that 6.1a forms an ion pair with the boronic acid. The resulting allylic cation then reacts with the diene to afford 6.1c.
A final example involves a report from Zhou in 2018, using a photocatalyst to induce a (4+3)-annulation and generate fluorinated benzoxepines (Scheme 6.2).[114] Based on their previous work with visible-light mediated (3+3)-annulations, they developed a dual C–F bond cleavage to construct a mono-fluorinated heterocycle using trifluoromethyl alkenes and o-hydroxyphenylacetic acid derivatives.[115] 1,3-Dicyano-2,4,5,6-tetrakis(N,N-diphenylamino)-benzene (4DPAIPN) was used as an alternative to the more expensive Ru and Ir photocatalysts. Numerous examples were reported with good to excellent yields. Halides, o-substituents and base-sensitive groups were tolerated.

Treatment with Li2CO3 gave significant amounts of the intermediate 6.2d′. This intermediate was subjected to the reaction conditions in the absence of light or a photocatalyst and the desired (4+3)-cycloadduct 6.2c′ was formed in 97% yield, supporting the fact that the photocatalyst and visible light were only required for the formation of 6.2d.
They proposed a mechanism wherein 6.2a is deprotonated to 6.2e, which, in comparison to 6.2a, has a lower oxidation potential. Upon irradiation, the excited 4DPAIPN* accepts an electron from 6.2e to generate a radical, which subsequently decarboxylates to afford benzylic radical 6.2f. The benzyl radical adds to alkene 6.2b to give 6.2g. Single-electron reduction by DPAIPN– gives radical anion 6.2h, which undergoes a β-F elimination to give 6.2d, followed by a rapid SNV reaction to give the fluorinated dihydrobenzoxepine 6.2c.
7
Conclusion
In conclusion, there are numerous pathways by which (4+3)-annulations occur. Many involve base-mediated procedures. NHC activation with α,β-unsaturated aldehydes, or enantioselective processes using chiral phosphoric acids, are of particular interest. This latter approach is frequently used to generate enantioenriched products from stable substrates.
Methods involving Lewis acids, bases, amines, and phosphines often give racemic products and require stoichiometric amounts of the reagent. Further investigation could enable catalytic and enantioselective transformations to furnish complex, enantioenriched cycloadducts.
New results using boronic acids or photocatalysts are emerging. For example, the use of boronic acid catalysis as an alternative to Lewis or Brønsted acid catalysis could be very useful. We can anticipate further advances using these approaches in the coming years.
-
References
- 1 Hu F, Ng J, Chiu P. Synthesis 2019; 51: 1073
- 2 Harmata M. Chem. Commun. 2010; 46: 8886
- 3 Harmata M. Tetrahedron Lett. 1997; 53: 6235
- 4 Fernández I, Mascareñas JL. Org. Biomol. Chem. 2012; 10: 699
- 5 Yin Z, He Y, Chiu P. Chem. Soc. Rev. 2018; 47: 8881
- 6 Selvaraj K, Chauhan S, Sandeep K, Swamy KC. K. Chem. Asian J. 2020; 15: 2380
- 7 Lam H, Lautens M. Synthesis 2020; 52: 2427
- 8 Feng J, Zhou M, Lin X, Lu A, Zhang X, Zhao M. Org. Lett. 2019; 21: 6245
- 9 Jeffrey CS, Barnes KL, Eickhoff JA, Carson CR. J. Am. Chem. Soc. 2011; 133: 7688
- 10 Jeffrey CS, Anumandla D, Carson CR. Org. Lett. 2012; 14: 5764
- 11 Son EC, Lee J, Kim S.-G. Eur. J. Org. Chem. 2020; 3090
- 12 Li W, Wei J, Jia Q, Du Z, Zhang K, Wang J. Chem. Eur. J. 2014; 20: 6592
- 13 Hesping L, Biswas A, Daniliuc CG, Mück-Lichtenfeld C, Studer A. Chem. Sci. 2015; 6: 1252
- 14 Gao Z.-H, Chen X.-Y, Cheng J.-T, Liao W.-L, Ye S. Chem. Commun. 2015; 51: 9328
- 15 Zhou Y.-Y, Li J, Ling L, Liao S.-H, Sun X.-L, Li Y.-X, Wang L.-J, Tang Y. Angew. Chem. Int. Ed. 2013; 52: 1452
- 16 Zhang L, Liu H, Qiao G, Hou Z, Liu Y, Xiao Y, Guo H. J. Am. Chem. Soc. 2015; 137: 4316
- 17 Li Z, Yu H, Liu Y, Zhou L, Sun Z, Guo H. Adv. Synth. Catal. 2016; 358: 1880
- 18 Xu J, Yuan S, Peng J, Miao M, Chen Z, Ren H. Org. Biomol. Chem. 2017; 15: 7513
- 19 Hu X.-Q, Chen J.-R, Gao S, Feng B, Lu L.-Q, Xiao W.-J. Chem. Commun. 2013; 49: 7905
- 20 Li Z, Li S, Kan T, Wang X, Xin X, Hou Y, Gong P. Adv. Synth. Catal. 2020; 362: 2626
- 21 Zhao H.-W, Pang H.-L, Tian T, Li B, Chen X.-Q, Song X.-Q, Meng W, Yang Z, Liu Y.-Y, Zhao Y.-D. Adv. Synth. Catal. 2016; 358: 1826
- 22 Zheng Y.-S, Tu L, Gao L.-M, Huang R, Feng T, Sun H, Wang W.-X, Li Z.-H, Liu J.-K. Org. Biomol. Chem. 2018; 16: 2639
- 23 Zhang X, Pan Y, Liang P, Pang L, Ma X, Jiao W, Shao H. Adv. Synth. Catal. 2018; 360: 3015
- 24 Guo Z, Jia H, Liu H, Wang Q, Huang J, Guo H. Org. Lett. 2018; 20: 2939
- 25 Rolfe A, Samarakoon TB, Hanson PR. Org. Lett. 2010; 12: 1216
- 26 Cleator E, Baxter CA, O’Hagan M, O’Riordan TJ. C, Sheen FJ, Stewart GW. Tetrahedron Lett. 2010; 51: 1079
- 27 Albanese D, Landini D, Lupi V, Penso M. Ind. Eng. Chem. Res. 2003; 42: 680
- 28 Albanese D, Landini D, Penso M. Chem. Commun. 1999; 2095
- 29 Hu J, Liu Y, Gong Y. Adv. Synth. Catal. 2015; 357: 2781
- 30 Cera G, Piscitelli S, Chiarucci M, Fabrizi G, Goggiamani A, Ramón RS, Nolan SP, Bandini M. Angew. Chem. Int. Ed. 2012; 51: 9891
- 31 Lee HS, Kim SH, Kim TH, Kim JN. Tetrahedron Lett. 2008; 49: 1773
- 32 England DB, Padwa A. J. Org. Chem. 2008; 73: 2792
- 33 England DB, Padwa A. Org. Lett. 2007; 9: 3249
- 34 Pérez-Serrano L, Casarrubios L, Domínguez G, Pérez-Castells J. Chem. Commun. 2001; 2602
- 35 Pérez-Serrano L, Domínguez G, Pérez-Castells J. J. Org. Chem. 2004; 69: 5413
- 36 Suryavanshi PA, Sridharan V, Menéndez JC. Org. Biomol. Chem. 2010; 8: 3426
- 37 Kusama H, Suzuki Y, Takaya J, Iwasawa N. Org. Lett. 2006; 8: 895
- 38 Zhang Y, Yu A, Jia J, Ma S, Li K, Wei Y, Meng X. Chem. Commun. 2017; 53: 10672
- 39 Li J.-L, Dai Q.-S, Yang K.-C, Liu Y, Zhang X, Leng H.-J, Peng C, Huang W, Li Q.-Z. Org. Lett. 2018; 20: 7628
- 40 Dinda BK, Jana AK, Mal D. Chem. Commun. 2012; 48: 3999
- 41 Ouyang L, Qi C, He H, Peng Y, Xiong W, Ren Y, Jiang H. J. Org. Chem. 2016; 81: 912
- 42 Chen L.-L, Zhang J.-W, Chen P, Zhang S, Yang W.-W, Fu J.-Y, Zhu J.-Y, Wang Y.-B. Org. Lett. 2019; 21: 5457
- 43 Chen Q, Wang Y, Hua R. Molecules 2019; 24: 3773
- 44 Jones DE, Harmata M. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses. Nishiwaki N. John Wiley & Sons; Hoboken: 2014
- 45 Jones DE, Harmata M. In Metal-Catalyzed Cyclization Reactions 2, Science of Synthesis . Ma S, Gao S. Georg Thieme Verlag; Stuttgart: 2016: 319
- 46 Dennis N, Ibrahim B, Katritzky AR. J. Chem. Soc., Perkin Trans. 1 1976; 2307
- 47 Fu C, Lora N, Kirchhoefer PL, Lee DR, Altenhofer E, Barnes CL, Hungerford NL, Krenske EH, Harmata M. Angew. Chem. Int. Ed. 2017; 56: 14682
- 48 Lv H, Jia W.-Q, Sun L.-H, Ye S. Angew. Chem. Int. Ed. 2013; 52: 8607
- 49 Liang Z.-Q, Gao Z.-H, Jia W.-Q, Ye S. Chem. Eur. J. 2015; 21: 1868
- 50 Liang Z.-Q, Yi L, Chen K.-Q, Ye S. J. Org. Chem. 2016; 81: 4841
- 51 Chen K.-Q, Gao Z.-H, Ye S. Org. Chem. Front. 2019; 6: 405
- 52 Xia F, Chen X.-Y, Ye S. J. Org. Chem. 2018; 83: 15178
- 53 Fang C, Lu T, Zhu J, Sun K, Du D. Org. Lett. 2017; 19: 3470
- 54 Fang C, Cao J, Sun K, Zhu J, Lu T, Du D. Chem. Eur. J. 2018; 24: 2103
- 55 Fukata Y, Asano K, Matsubara S. J. Am. Chem. Soc. 2015; 137: 5320
- 56 Li W, Yuan H, Liu Z, Zhang Z, Cheng Y, Li P. Adv. Synth. Catal. 2018; 360: 2460
- 57 Wang M, Huang Z, Xu J, Chi YR. J. Am. Chem. Soc. 2014; 136: 1214
- 58 Izquierdo J, Orue A, Scheidt KA. J. Am. Chem. Soc. 2013; 135: 10634
- 59 Wang L, Li S, Blümel M, Philipps AR, Wang A, Puttreddy R, Rissanen K, Enders D. Angew. Chem. Int. Ed. 2016; 55: 11110
- 60 Guo C, Sahoo B, Daniliuc CG, Glorius F. J. Am. Chem. Soc. 2014; 136: 17402
- 61 Zhu S.-Y, Zhang Y, Wang W, Hui X.-P. Org. Lett. 2017; 19: 5380
- 62 Liu D, Hu Z, Zhang Y, Gong M, Fu Z, Huang W. Chem. Eur. J. 2019; 25: 11223
- 63 Guo H, Fan YC, Sun Z, Wu Y, Kwon O. Chem. Rev. 2018; 118: 10049
- 64 Wei Y, Shi M. In Organocatalytic Cycloadditions for Synthesis of Carbo- and Heterocycles, 1st ed. Shi M, Wei Y, Zhao M.-X, Zhang J. Wiley-VCH; Weinheim: 2018: 141
- 65 Na R, Jing C, Xu Q, Jiang H, Wu X, Shi J, Zhong J, Wang M, Benitez D, Tkatchouk E, Goddard WA, Guo H, Kwon O. J. Am. Chem. Soc. 2011; 133: 13337
- 66 Jing C, Na R, Wang B, Liu H, Zhang L, Liu J, Wang M, Zhong J, Kwon O, Guo H. Adv. Synth. Catal. 2012; 354: 1023
- 67 Yuan C, Zhou L, Xia M, Sun Z, Wang D, Guo H. Org. Lett. 2016; 18: 5644
- 68 Kumar K, Kapoor R, Kapur A, Ishar MP. S. Org. Lett. 2000; 2: 2023
- 69 Zheng S, Lu X. Org. Lett. 2009; 11: 3978
- 70 Hillman ME. D, Baylis AB. (Celanese Corp, USA) US3743669A 1973
- 71 Chen J, Huang Y. Org. Lett. 2017; 19: 5609
- 72 Chen J, Yin Z, Huang Y. Org. Lett. 2019; 21: 7060
- 73 Zhan G, Shi M.-L, He Q, Du W, Chen Y.-C. Org. Lett. 2015; 17: 4750
- 74 Ivanov KL, Melnikov MY, Budynina EM. Org. Lett. 2019; 21: 4464
- 75 Lewis Acid Reagents: A Practical Approach . Yamamoto H. Oxford University Press; Oxford: 1999
- 76 Lewis Acids in Organic Synthesis . Yamamoto H. Wiley-VCH; Weinheim: 2000
- 77 Pathipati SR, Eriksson L, Selander N. Chem. Commun. 2017; 53: 11353
- 78 Han X, Li H, Hughes RP, Wu J. Angew. Chem. Int. Ed. 2012; 51: 10390
- 79 Wani IA, Bhattacharyya A, Sayyad M, Ghorai MK. Org. Biomol. Chem. 2018; 16: 2910
- 80 Song B, Li L.-H, Song X.-R, Qiu Y.-F, Zhong M.-J, Zhou P.-X, Liang Y.-M. Chem. Eur. J. 2014; 20: 5910
- 81 Hoffmann R, Stohrer WD. J. Am. Chem. Soc. 1971; 93: 6941
- 82 Dewar MJ. S, Wade LE. J. Am. Chem. Soc. 1977; 99: 4417
- 83 Han Y.-P, Song X.-R, Qiu Y.-F, Zhang H.-R, Li L.-H, Jin D.-P, Sun X.-Q, Liu X.-Y, Liang Y.-M. Org. Lett. 2016; 18: 940
- 84 Shu C, Wang Y.-H, Zhou B, Li X.-L, Ping Y.-F, Lu X, Ye L.-W. J. Am. Chem. Soc. 2015; 137: 9567
- 85 Siyang HX, Ji XY, Wu XR, Wu XY, Liu PN. Org. Lett. 2015; 17: 5220
- 86 Stevens AC, Palmer C, Pagenkopf BL. Org. Lett. 2011; 13: 1528
- 87 Zhang C, Tian J, Ren J, Wang Z. Chem. Eur. J. 2017; 23: 1231
- 88 Garve LK. B, Jones PG, Werz DB. Angew. Chem. Int. Ed. 2017; 56: 9226
- 89 Augustin AU, Merz JL, Jones PG, Mlostoń G, Werz DB. Org. Lett. 2019; 21: 9405
- 90 Morgan TD. R, LeFort FM, Li Z, Marx VM, Boyd RJ, Burnell DJ. Eur. J. Org. Chem. 2015; 2952
- 91 Nazarov IN, Zaretskaya I. Izv. Akad. Nauk. SSSR Ser. Khim. 1941; 211
- 92 Habermas KL, Denmark SE, Jones TK. Org. React. 1994; 45: 1
- 93 Winne JM, Catak S, Waroquier M, Van Speybroeck V. Angew. Chem. Int. Ed. 2011; 50: 11990
- 94 Laplace DR, Verbraeken B, Van Hecke K, Winne JM. Chem. Eur. J. 2014; 20: 253
- 95 Zong Y, Wang W, Xu T. Mar. Drugs 2018; 16: 115
- 96 Marion F, Williams DE, Patrick BO, Hollander I, Mallon R, Kim SC, Roll DM, Feldberg L, Van Soest R, Andersen RJ. Org. Lett. 2006; 8: 321
- 97 Xu J, Rawal VH. J. Am. Chem. Soc. 2019; 141: 4820
- 98 Hullaert J, Denoo B, Christiaens M, Callebaut B, Winne JM. Synlett 2017; 28: 2345
- 99 Pattenden G, Winne JM. Tetrahedron Lett. 2009; 50: 7310
- 100 Christiaens M, Hullaert J, Van Hecke K, Laplace D, Winne JM. Chem. Eur. J. 2018; 24: 13783
- 101 Winne J, Laplace D. Synlett 2015; 26: 467
- 102 Callebaut B, Hullaert J, Hecke KV, Winne JM. Org. Lett. 2019; 21: 310
- 103 Zhang J, Li L, Wang Y, Wang W, Xue J, Li Y. Org. Lett. 2012; 14: 4528
- 104 George JH, Baldwin JE, Adlington RM. Org. Lett. 2010; 12: 2394
- 105 Zhang H.-H, Zhu Z.-Q, Fan T, Liang J, Shi F. Adv. Synth. Catal. 2016; 358: 1259
- 106 Lam H, Qureshi Z, Wegmann M, Lautens M. Angew. Chem. Int. Ed. 2018; 57: 16185
- 107 Sun M, Ma C, Zhou SJ, Lou SF, Xiao J, Jiao Y, Shi F. Angew. Chem. Int. Ed. 2019; 58: 8703
- 108 Gelis C, Levitre G, Merad J, Retailleau P, Neuville L, Masson G. Angew. Chem. Int. Ed. 2018; 57: 12121
- 109 Mei GJ, Zhu ZQ, Zhao JJ, Bian CY, Chen J, Chen RW, Shi F. Chem. Commun. 2017; 53: 2768
- 110 Li C, Wang CS, Li TZ, Mei GJ, Shi F. Org. Lett. 2019; 21: 598
- 111 Cao K.-S, Bian H.-X, Zheng W.-H. Org. Biomol. Chem. 2015; 13: 6449
- 112 He S, Hsung RP, Presser WR, Ma Z.-X, Haugen BJ. Org. Lett. 2014; 16: 2180
- 113 Zhang Z, Li L, Wang Y, Wang W, Xue J, Li Y. Org. Lett. 2012; 14: 4528
- 114 Chen H, He Y, Zhou L. Org. Chem. Front. 2018; 5: 3240
- 115 Li L, Xiao T, Chen H, Zhou L. Chem. Eur. J. 2017; 23: 2249
Corresponding Authors
Publication History
Received: 10 January 2021
Accepted after revision: 11 January 2021
Article published online:
23 February 2021
© 2021. Thieme. All rights reserved
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Hu F, Ng J, Chiu P. Synthesis 2019; 51: 1073
- 2 Harmata M. Chem. Commun. 2010; 46: 8886
- 3 Harmata M. Tetrahedron Lett. 1997; 53: 6235
- 4 Fernández I, Mascareñas JL. Org. Biomol. Chem. 2012; 10: 699
- 5 Yin Z, He Y, Chiu P. Chem. Soc. Rev. 2018; 47: 8881
- 6 Selvaraj K, Chauhan S, Sandeep K, Swamy KC. K. Chem. Asian J. 2020; 15: 2380
- 7 Lam H, Lautens M. Synthesis 2020; 52: 2427
- 8 Feng J, Zhou M, Lin X, Lu A, Zhang X, Zhao M. Org. Lett. 2019; 21: 6245
- 9 Jeffrey CS, Barnes KL, Eickhoff JA, Carson CR. J. Am. Chem. Soc. 2011; 133: 7688
- 10 Jeffrey CS, Anumandla D, Carson CR. Org. Lett. 2012; 14: 5764
- 11 Son EC, Lee J, Kim S.-G. Eur. J. Org. Chem. 2020; 3090
- 12 Li W, Wei J, Jia Q, Du Z, Zhang K, Wang J. Chem. Eur. J. 2014; 20: 6592
- 13 Hesping L, Biswas A, Daniliuc CG, Mück-Lichtenfeld C, Studer A. Chem. Sci. 2015; 6: 1252
- 14 Gao Z.-H, Chen X.-Y, Cheng J.-T, Liao W.-L, Ye S. Chem. Commun. 2015; 51: 9328
- 15 Zhou Y.-Y, Li J, Ling L, Liao S.-H, Sun X.-L, Li Y.-X, Wang L.-J, Tang Y. Angew. Chem. Int. Ed. 2013; 52: 1452
- 16 Zhang L, Liu H, Qiao G, Hou Z, Liu Y, Xiao Y, Guo H. J. Am. Chem. Soc. 2015; 137: 4316
- 17 Li Z, Yu H, Liu Y, Zhou L, Sun Z, Guo H. Adv. Synth. Catal. 2016; 358: 1880
- 18 Xu J, Yuan S, Peng J, Miao M, Chen Z, Ren H. Org. Biomol. Chem. 2017; 15: 7513
- 19 Hu X.-Q, Chen J.-R, Gao S, Feng B, Lu L.-Q, Xiao W.-J. Chem. Commun. 2013; 49: 7905
- 20 Li Z, Li S, Kan T, Wang X, Xin X, Hou Y, Gong P. Adv. Synth. Catal. 2020; 362: 2626
- 21 Zhao H.-W, Pang H.-L, Tian T, Li B, Chen X.-Q, Song X.-Q, Meng W, Yang Z, Liu Y.-Y, Zhao Y.-D. Adv. Synth. Catal. 2016; 358: 1826
- 22 Zheng Y.-S, Tu L, Gao L.-M, Huang R, Feng T, Sun H, Wang W.-X, Li Z.-H, Liu J.-K. Org. Biomol. Chem. 2018; 16: 2639
- 23 Zhang X, Pan Y, Liang P, Pang L, Ma X, Jiao W, Shao H. Adv. Synth. Catal. 2018; 360: 3015
- 24 Guo Z, Jia H, Liu H, Wang Q, Huang J, Guo H. Org. Lett. 2018; 20: 2939
- 25 Rolfe A, Samarakoon TB, Hanson PR. Org. Lett. 2010; 12: 1216
- 26 Cleator E, Baxter CA, O’Hagan M, O’Riordan TJ. C, Sheen FJ, Stewart GW. Tetrahedron Lett. 2010; 51: 1079
- 27 Albanese D, Landini D, Lupi V, Penso M. Ind. Eng. Chem. Res. 2003; 42: 680
- 28 Albanese D, Landini D, Penso M. Chem. Commun. 1999; 2095
- 29 Hu J, Liu Y, Gong Y. Adv. Synth. Catal. 2015; 357: 2781
- 30 Cera G, Piscitelli S, Chiarucci M, Fabrizi G, Goggiamani A, Ramón RS, Nolan SP, Bandini M. Angew. Chem. Int. Ed. 2012; 51: 9891
- 31 Lee HS, Kim SH, Kim TH, Kim JN. Tetrahedron Lett. 2008; 49: 1773
- 32 England DB, Padwa A. J. Org. Chem. 2008; 73: 2792
- 33 England DB, Padwa A. Org. Lett. 2007; 9: 3249
- 34 Pérez-Serrano L, Casarrubios L, Domínguez G, Pérez-Castells J. Chem. Commun. 2001; 2602
- 35 Pérez-Serrano L, Domínguez G, Pérez-Castells J. J. Org. Chem. 2004; 69: 5413
- 36 Suryavanshi PA, Sridharan V, Menéndez JC. Org. Biomol. Chem. 2010; 8: 3426
- 37 Kusama H, Suzuki Y, Takaya J, Iwasawa N. Org. Lett. 2006; 8: 895
- 38 Zhang Y, Yu A, Jia J, Ma S, Li K, Wei Y, Meng X. Chem. Commun. 2017; 53: 10672
- 39 Li J.-L, Dai Q.-S, Yang K.-C, Liu Y, Zhang X, Leng H.-J, Peng C, Huang W, Li Q.-Z. Org. Lett. 2018; 20: 7628
- 40 Dinda BK, Jana AK, Mal D. Chem. Commun. 2012; 48: 3999
- 41 Ouyang L, Qi C, He H, Peng Y, Xiong W, Ren Y, Jiang H. J. Org. Chem. 2016; 81: 912
- 42 Chen L.-L, Zhang J.-W, Chen P, Zhang S, Yang W.-W, Fu J.-Y, Zhu J.-Y, Wang Y.-B. Org. Lett. 2019; 21: 5457
- 43 Chen Q, Wang Y, Hua R. Molecules 2019; 24: 3773
- 44 Jones DE, Harmata M. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses. Nishiwaki N. John Wiley & Sons; Hoboken: 2014
- 45 Jones DE, Harmata M. In Metal-Catalyzed Cyclization Reactions 2, Science of Synthesis . Ma S, Gao S. Georg Thieme Verlag; Stuttgart: 2016: 319
- 46 Dennis N, Ibrahim B, Katritzky AR. J. Chem. Soc., Perkin Trans. 1 1976; 2307
- 47 Fu C, Lora N, Kirchhoefer PL, Lee DR, Altenhofer E, Barnes CL, Hungerford NL, Krenske EH, Harmata M. Angew. Chem. Int. Ed. 2017; 56: 14682
- 48 Lv H, Jia W.-Q, Sun L.-H, Ye S. Angew. Chem. Int. Ed. 2013; 52: 8607
- 49 Liang Z.-Q, Gao Z.-H, Jia W.-Q, Ye S. Chem. Eur. J. 2015; 21: 1868
- 50 Liang Z.-Q, Yi L, Chen K.-Q, Ye S. J. Org. Chem. 2016; 81: 4841
- 51 Chen K.-Q, Gao Z.-H, Ye S. Org. Chem. Front. 2019; 6: 405
- 52 Xia F, Chen X.-Y, Ye S. J. Org. Chem. 2018; 83: 15178
- 53 Fang C, Lu T, Zhu J, Sun K, Du D. Org. Lett. 2017; 19: 3470
- 54 Fang C, Cao J, Sun K, Zhu J, Lu T, Du D. Chem. Eur. J. 2018; 24: 2103
- 55 Fukata Y, Asano K, Matsubara S. J. Am. Chem. Soc. 2015; 137: 5320
- 56 Li W, Yuan H, Liu Z, Zhang Z, Cheng Y, Li P. Adv. Synth. Catal. 2018; 360: 2460
- 57 Wang M, Huang Z, Xu J, Chi YR. J. Am. Chem. Soc. 2014; 136: 1214
- 58 Izquierdo J, Orue A, Scheidt KA. J. Am. Chem. Soc. 2013; 135: 10634
- 59 Wang L, Li S, Blümel M, Philipps AR, Wang A, Puttreddy R, Rissanen K, Enders D. Angew. Chem. Int. Ed. 2016; 55: 11110
- 60 Guo C, Sahoo B, Daniliuc CG, Glorius F. J. Am. Chem. Soc. 2014; 136: 17402
- 61 Zhu S.-Y, Zhang Y, Wang W, Hui X.-P. Org. Lett. 2017; 19: 5380
- 62 Liu D, Hu Z, Zhang Y, Gong M, Fu Z, Huang W. Chem. Eur. J. 2019; 25: 11223
- 63 Guo H, Fan YC, Sun Z, Wu Y, Kwon O. Chem. Rev. 2018; 118: 10049
- 64 Wei Y, Shi M. In Organocatalytic Cycloadditions for Synthesis of Carbo- and Heterocycles, 1st ed. Shi M, Wei Y, Zhao M.-X, Zhang J. Wiley-VCH; Weinheim: 2018: 141
- 65 Na R, Jing C, Xu Q, Jiang H, Wu X, Shi J, Zhong J, Wang M, Benitez D, Tkatchouk E, Goddard WA, Guo H, Kwon O. J. Am. Chem. Soc. 2011; 133: 13337
- 66 Jing C, Na R, Wang B, Liu H, Zhang L, Liu J, Wang M, Zhong J, Kwon O, Guo H. Adv. Synth. Catal. 2012; 354: 1023
- 67 Yuan C, Zhou L, Xia M, Sun Z, Wang D, Guo H. Org. Lett. 2016; 18: 5644
- 68 Kumar K, Kapoor R, Kapur A, Ishar MP. S. Org. Lett. 2000; 2: 2023
- 69 Zheng S, Lu X. Org. Lett. 2009; 11: 3978
- 70 Hillman ME. D, Baylis AB. (Celanese Corp, USA) US3743669A 1973
- 71 Chen J, Huang Y. Org. Lett. 2017; 19: 5609
- 72 Chen J, Yin Z, Huang Y. Org. Lett. 2019; 21: 7060
- 73 Zhan G, Shi M.-L, He Q, Du W, Chen Y.-C. Org. Lett. 2015; 17: 4750
- 74 Ivanov KL, Melnikov MY, Budynina EM. Org. Lett. 2019; 21: 4464
- 75 Lewis Acid Reagents: A Practical Approach . Yamamoto H. Oxford University Press; Oxford: 1999
- 76 Lewis Acids in Organic Synthesis . Yamamoto H. Wiley-VCH; Weinheim: 2000
- 77 Pathipati SR, Eriksson L, Selander N. Chem. Commun. 2017; 53: 11353
- 78 Han X, Li H, Hughes RP, Wu J. Angew. Chem. Int. Ed. 2012; 51: 10390
- 79 Wani IA, Bhattacharyya A, Sayyad M, Ghorai MK. Org. Biomol. Chem. 2018; 16: 2910
- 80 Song B, Li L.-H, Song X.-R, Qiu Y.-F, Zhong M.-J, Zhou P.-X, Liang Y.-M. Chem. Eur. J. 2014; 20: 5910
- 81 Hoffmann R, Stohrer WD. J. Am. Chem. Soc. 1971; 93: 6941
- 82 Dewar MJ. S, Wade LE. J. Am. Chem. Soc. 1977; 99: 4417
- 83 Han Y.-P, Song X.-R, Qiu Y.-F, Zhang H.-R, Li L.-H, Jin D.-P, Sun X.-Q, Liu X.-Y, Liang Y.-M. Org. Lett. 2016; 18: 940
- 84 Shu C, Wang Y.-H, Zhou B, Li X.-L, Ping Y.-F, Lu X, Ye L.-W. J. Am. Chem. Soc. 2015; 137: 9567
- 85 Siyang HX, Ji XY, Wu XR, Wu XY, Liu PN. Org. Lett. 2015; 17: 5220
- 86 Stevens AC, Palmer C, Pagenkopf BL. Org. Lett. 2011; 13: 1528
- 87 Zhang C, Tian J, Ren J, Wang Z. Chem. Eur. J. 2017; 23: 1231
- 88 Garve LK. B, Jones PG, Werz DB. Angew. Chem. Int. Ed. 2017; 56: 9226
- 89 Augustin AU, Merz JL, Jones PG, Mlostoń G, Werz DB. Org. Lett. 2019; 21: 9405
- 90 Morgan TD. R, LeFort FM, Li Z, Marx VM, Boyd RJ, Burnell DJ. Eur. J. Org. Chem. 2015; 2952
- 91 Nazarov IN, Zaretskaya I. Izv. Akad. Nauk. SSSR Ser. Khim. 1941; 211
- 92 Habermas KL, Denmark SE, Jones TK. Org. React. 1994; 45: 1
- 93 Winne JM, Catak S, Waroquier M, Van Speybroeck V. Angew. Chem. Int. Ed. 2011; 50: 11990
- 94 Laplace DR, Verbraeken B, Van Hecke K, Winne JM. Chem. Eur. J. 2014; 20: 253
- 95 Zong Y, Wang W, Xu T. Mar. Drugs 2018; 16: 115
- 96 Marion F, Williams DE, Patrick BO, Hollander I, Mallon R, Kim SC, Roll DM, Feldberg L, Van Soest R, Andersen RJ. Org. Lett. 2006; 8: 321
- 97 Xu J, Rawal VH. J. Am. Chem. Soc. 2019; 141: 4820
- 98 Hullaert J, Denoo B, Christiaens M, Callebaut B, Winne JM. Synlett 2017; 28: 2345
- 99 Pattenden G, Winne JM. Tetrahedron Lett. 2009; 50: 7310
- 100 Christiaens M, Hullaert J, Van Hecke K, Laplace D, Winne JM. Chem. Eur. J. 2018; 24: 13783
- 101 Winne J, Laplace D. Synlett 2015; 26: 467
- 102 Callebaut B, Hullaert J, Hecke KV, Winne JM. Org. Lett. 2019; 21: 310
- 103 Zhang J, Li L, Wang Y, Wang W, Xue J, Li Y. Org. Lett. 2012; 14: 4528
- 104 George JH, Baldwin JE, Adlington RM. Org. Lett. 2010; 12: 2394
- 105 Zhang H.-H, Zhu Z.-Q, Fan T, Liang J, Shi F. Adv. Synth. Catal. 2016; 358: 1259
- 106 Lam H, Qureshi Z, Wegmann M, Lautens M. Angew. Chem. Int. Ed. 2018; 57: 16185
- 107 Sun M, Ma C, Zhou SJ, Lou SF, Xiao J, Jiao Y, Shi F. Angew. Chem. Int. Ed. 2019; 58: 8703
- 108 Gelis C, Levitre G, Merad J, Retailleau P, Neuville L, Masson G. Angew. Chem. Int. Ed. 2018; 57: 12121
- 109 Mei GJ, Zhu ZQ, Zhao JJ, Bian CY, Chen J, Chen RW, Shi F. Chem. Commun. 2017; 53: 2768
- 110 Li C, Wang CS, Li TZ, Mei GJ, Shi F. Org. Lett. 2019; 21: 598
- 111 Cao K.-S, Bian H.-X, Zheng W.-H. Org. Biomol. Chem. 2015; 13: 6449
- 112 He S, Hsung RP, Presser WR, Ma Z.-X, Haugen BJ. Org. Lett. 2014; 16: 2180
- 113 Zhang Z, Li L, Wang Y, Wang W, Xue J, Li Y. Org. Lett. 2012; 14: 4528
- 114 Chen H, He Y, Zhou L. Org. Chem. Front. 2018; 5: 3240
- 115 Li L, Xiao T, Chen H, Zhou L. Chem. Eur. J. 2017; 23: 2249